聚氨基酸、其制备方法及应用与流程
本发明涉及聚合物技术领域,尤其涉及聚氨基酸、其制备方法及应用。
背景技术:
近些年来由于多肽可通过氨基酸的复合和特定序列而形成特异性的二级结构或纳米结构,使其被广泛的应用于药物传递载体、组织工程支架等生物领域。氨基酸具有特异性的手性结构,可以分为天然的l-氨基酸和非天然的d-氨基酸。多肽作为生物制药中间体或疫苗的应用基于立体构型之间化学或酶的转化。从生物学角度来说,可以通过改变其构型来改变其水解和降解。其次,温度敏感聚合物随着温度的增加经历溶胶-凝胶的转变,尤其是在人体温37℃下。相转变聚合物已经被广泛用于药物传递载体、可注射组织工程支架以及术后防黏连生物材料。
基于以上,科研工作者一直致力于研究水凝胶的应用。其中,肿瘤疫苗的载药方式的研究更能引起关注。由于聚合物能够在水中形成温度敏感的材料,随着温度的增加形成凝胶包载药物等,且聚合物纳米凝胶具有毒性低,治疗效果好,温度环境决定成胶性能等优点。
技术实现要素:
有鉴于此,本发明要解决的技术问题在于提供一种聚氨基酸、其制备方法及应用,采用本发明的聚氨基酸得到的聚氨基酸水凝胶在体内能够引起较大的炎症反应。
本发明提供了一种聚氨基酸,具有式(ⅰ)或式(ⅱ)所示结构:
其中,r为-h、-ch3、(ch3)2ch-或ch3)2chch2-;
30≤m≤200,4≤n≤50。
优选的,60≤m≤190,10≤n≤40。
本发明还提供了一种聚氨基酸的制备方法,包括以下步骤:
将具有式(ⅲ)结构的端氨基聚乙二醇单甲醚和l-氨基酸-n-内羧酸酐在第一溶剂中反应,得到具有式(ⅰ)所示结构的聚氨基酸;所述l-氨基酸-n-内羧酸酐为l-丙氨酸-n-内羧酸酐、l-缬氨酸-n-内羧酸酐、l-甘氨酸-n-内羧酸酐或l-亮氨酸-n-内羧酸酐;
式(ⅰ)中,r为-h、-ch3、(ch3)2ch-或ch3)2chch2-;
30≤m≤200,4≤n≤50;
式(ⅲ)中,30≤i≤200;
或
将所述具有式(ⅲ)结构的端氨基聚乙二醇单甲醚与d-氨基酸-n-内羧酸酐在第一溶剂中反应,得到具有式(ⅱ)所示结构的聚氨基酸;所述d-氨基酸-n-内羧酸酐为d-缬氨酸-n-内羧酸酐、d-丙氨酸-n-内羧酸酐、d-甘氨酸-n-内羧酸酐或d-亮氨酸-n-内羧酸酐;
式(ⅱ)中,r为-h、-ch3、(ch3)2ch-或ch3)2chch2-;
30≤m≤200,4≤n≤50。
优选的,所述具有式(ⅲ)结构的端氨基聚乙二醇单甲醚和l-氨基酸-n-内羧酸酐的摩尔比为1:1~30;
所述具有式(ⅲ)结构的端氨基聚乙二醇单甲醚与d-氨基酸-n-内羧酸酐的摩尔比为1:1~30;
所述第一溶剂为n,n-二甲基甲酰胺。
优选的,所述具有式(ⅲ)结构的端氨基聚乙二醇单甲醚按照以下方法进行制备:
a)将聚乙二醇单甲醚、三乙胺和甲基磺酰氯在第二溶剂中进行反应,得到中间产物;
b)将所述中间产物与氨水反应,得到具有式(ⅲ)结构的端氨基聚乙二醇单甲醚。
优选的,步骤a)中,所述聚乙二醇单甲醚、三乙胺和甲基磺酰氯的摩尔比为1:1.5~10:10~39;
所述第二溶剂为二氯甲烷;
所述反应的温度为10~40℃,所述反应的时间为3~8d。
优选的,步骤b)中,所述中间产物与氨水的质量比为1:10~40;
所述反应的温度为10~40℃,所述反应的时间为3~7d。
优选的,所述反应的温度为4~85℃,所述反应的时间为60~100h。
本发明还提供了一种聚氨基酸水凝胶,由上文所述的聚氨基酸和溶剂组成;或由上文所述制备方法制得的聚氨基酸和溶剂组成。
本发明还提供了一种载药凝胶,包括聚氨基酸和负载在聚氨基酸上的抗肿瘤药物,所述聚氨基酸为上文所述的聚氨基酸或上文所述制备方法制得的聚氨基酸。
本发明提供了一种聚氨基酸,具有式(ⅰ)或式(ⅱ)所示结构,其中,r为-h、-ch3、(ch3)2ch-或ch3)2chch2-;30≤m≤200,4≤n≤50。本申请提供的聚氨基酸一端是亲水的聚乙二醇单甲醚,另一端分别是疏水的聚-l-丙氨酸、聚-l-缬氨酸、聚-l-甘氨酸、聚-l-亮氨酸、聚-d-丙氨酸、聚-d-缬氨酸、聚-d-甘氨酸或聚-d-亮氨酸,该种具有亲水链段和疏水链段的两亲性聚氨基酸在水中能够形成纳米尺寸的物理凝胶,且在一定温度下能够成胶,因此该种物理凝胶能够作为药物传输和控制释放的载体材料。将本发明提供的聚氨基酸的水凝胶经皮下注射后,会引起较大的炎症反应,炎症反应可以通过产生一系列炎症因子刺激t细胞、dc细胞等产生一系列免疫响应,从而达到免疫治疗的效果。另外,本发明的聚氨基酸的成胶性能可通过温度、ph、光与电等实现可调控性,因此,本申请提供的水凝胶具有温度响应性。此外,本申请提供的载药凝胶中含有聚氨基酸,使该种载药凝胶具有较好的生物相容性、低毒性和生物降解性。
附图说明
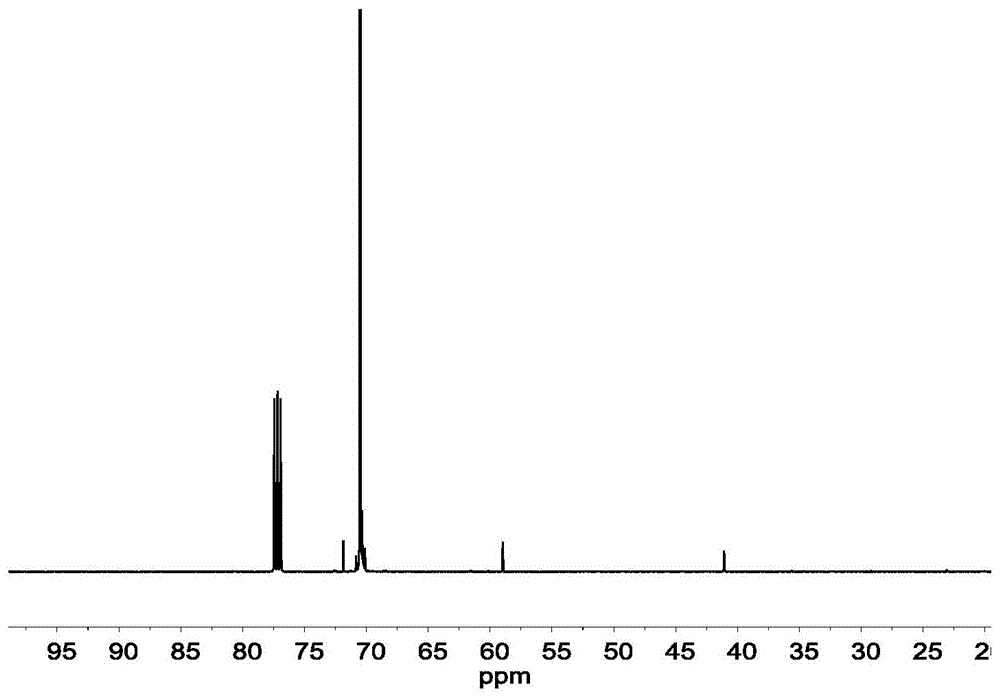
图1为本发明实施例1得到的端氨基聚乙二醇单甲醚的13c核磁谱图;
图2为本发明实施例12制备得到的聚氨基酸的1h核磁谱图;
图3为本发明实施例13制备得到的聚氨基酸的1h核磁谱图;
图4为本发明实施例16制备得到的聚氨基酸的1h核磁谱图;
图5为本发明实施例17制备得到的聚氨基酸的1h核磁谱图;
图6为本发明实施例20得到的凝胶的相图;
图7为本发明实施例21得到的凝胶的相图;
图8为本发明实施例13所得的聚-l-丙氨酸与实施例17所得的聚-d-丙氨酸的圆二色谱图;
图9为本发明实施例23得到的凝胶处组织的苏木精-伊红染色效果图;
图10为本发明实施例24得到的凝胶处组织的苏木精-伊红染色效果图;
图11为本发明实施例27得到的凝胶处组织的苏木精-伊红染色效果图;
图12为本发明实施例28得到的凝胶处组织的苏木精-伊红染色效果图。
具体实施方式
下面将结合本发明实施例,对本发明的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
本发明提供了一种聚氨基酸,具有式(ⅰ)或式(ⅱ)所示结构:
式(ⅰ)中,r为-h、-ch3、(ch3)2ch-或ch3)2chch2-;30≤m≤200,4≤n≤50。
本发明的实施例中,所述式(ⅰ)中,30≤m≤190;在某些实施例中,70≤m≤180;在某些实施例中,100≤m≤150、30≤m≤45或m=45。
在本发明的实施例中,所述式(ⅰ)中,5≤n≤33;在某些实施例中,10≤n≤20;在某些实施例中,12≤n≤18、n=17、n=27、n=33、n=15或n=32。
在本发明的实施例中,所述具有式(ⅰ)所示结构的聚氨基酸的数均分子量为2000~100000;在某些实施例中,所述具有式(ⅰ)所示结构的聚氨基酸的数均分子量为3000~90000;在某些实施例中,所述具有式(ⅰ)所示结构的聚氨基酸的数均分子量为4000~80000。
式(ⅱ)中,r为-h、-ch3、(ch3)2ch-或ch3)2chch2-;30≤m≤200,4≤n≤50。
本发明的实施例中,所述式(ⅱ)中,30≤m≤190;在某些实施例中,70≤m≤180;在某些实施例中,100≤m≤150。在本发明的实施例中,所述式(ⅱ)中,5≤n≤30;在某些实施例中,10≤n≤20;在某些实施例中,12≤n≤18。
在本发明的实施例中,所述具有式(ⅱ)所示结构的聚氨基酸的数均分子量为2000~100000;在某些实施例中,所述具有式(ⅱ)所示结构的聚氨基酸的数均分子量为3000~90000;在某些实施例中,所述具有式(ⅱ)所示结构的聚氨基酸的数均分子量为4000~80000。
本发明还提供了一种上文所述的聚氨基酸的制备方法,包括以下步骤:
将具有式(ⅲ)结构的端氨基聚乙二醇单甲醚和l-氨基酸-n-内羧酸酐在第一溶剂中反应,得到具有式(ⅰ)所示结构的聚氨基酸;所述l-氨基酸-n-内羧酸酐为l-丙氨酸-n-内羧酸酐、l-缬氨酸-n-内羧酸酐、l-甘氨酸-n-内羧酸酐或l-亮氨酸-n-内羧酸酐;
式(ⅰ)中,r为-h、-ch3、(ch3)2ch-或ch3)2chch2-;
30≤m≤200,4≤n≤50;
式(ⅲ)中,30≤i≤200;
或
将所述具有式(ⅲ)结构的端氨基聚乙二醇单甲醚与d-氨基酸-n-内羧酸酐在第一溶剂中反应,得到具有式(ⅱ)所示结构的聚氨基酸;所述d-氨基酸-n-内羧酸酐为d-缬氨酸-n-内羧酸酐、d-丙氨酸-n-内羧酸酐、d-甘氨酸-n-内羧酸酐或d-亮氨酸-n-内羧酸酐;
式(ⅱ)中,r为-h、-ch3、(ch3)2ch-或ch3)2chch2-;
30≤m≤200,4≤n≤50。
在上述第一种制备方法中,将具有式(ⅲ)结构的端氨基聚乙二醇单甲醚和l-氨基酸-n-内羧酸酐在第一溶剂中反应,得到具有式(ⅰ)所示结构的聚氨基酸。
具有式(ⅰ)所示结构的聚氨基酸的制备方法中,r、m和n的限定同上,在此不再赘述。式(ⅲ)中,30≤i≤200;在某些实施例中,60≤i≤190;在某些实施例中,70≤i≤180;在某些实施例中,100≤i≤150。
所述l-氨基酸-n-内羧酸酐为l-丙氨酸-n-内羧酸酐、l-缬氨酸-n-内羧酸酐、l-甘氨酸-n-内羧酸酐或l-亮氨酸-n-内羧酸酐。
在本发明的实施例中,所述l-缬氨酸-n-内羧酸酐具有式(ⅳ)所示的结构:
所述l-丙氨酸-n-内羧酸酐具有式(ⅴ)所示的结构:
所述l-甘氨酸-n-内羧酸酐具有式(ⅵ)所示的结构:
所述l-亮氨酸-n-内羧酸酐具有式(ⅶ)所示的结构:
在本发明的实施例中,所述第一溶剂为n,n-二甲基甲酰胺。
在本发明的实施例中,所述具有式(ⅲ)结构的端氨基聚乙二醇单甲醚按照以下方法进行制备:
a)将聚乙二醇单甲醚、三乙胺和甲基磺酰氯在第二溶剂中进行反应,得到中间产物;
b)将所述中间产物与氨水反应,得到具有式(ⅲ)结构的端氨基聚乙二醇单甲醚。
在本发明的实施例中,所述步骤a)为:向聚乙二醇单甲醚中依次加入第二溶剂、三乙胺和甲基磺酰氯进行反应,得到中间产物。在本发明的实施例中,聚乙二醇单甲醚中加入第二溶剂时的温度为55~75℃。在某些实施例中,聚乙二醇单甲醚中加入第二溶剂时的温度为60~70℃。在某些实施例中,所述三乙胺和甲基磺酰氯是在冰水浴的条件下加入的。
在本发明的实施例中,步骤a)中,所述反应的温度为10~40℃,所述反应的时间为3~8d。在某些实施例中,所述反应的温度为15~35℃。在某些实施例中,所述反应的温度为20~30℃。在某些实施例中,所述反应的时间为4~5d。在本发明的实施例中,所述反应在搅拌的条件下进行。在本发明的实施例中,所述反应在无水、无氧的条件下进行。
在本发明的实施例中,所述聚乙二醇单甲醚、三乙胺和甲基磺酰氯的摩尔比为1:1.5~10:10~39。在某些实施例中,所述聚乙二醇单甲醚、三乙胺和甲基磺酰氯的摩尔比为1:3.6:39、1:1.78:19.4或1:3.5:28.75。
本发明的实施例中,所述第二溶剂为二氯甲烷。本发明对所述第二溶剂的用量并无特殊的限制。在某些实施例中,所述聚乙二醇单甲醚和二氯甲烷的用量比为20g:200ml。
本发明的实施例中,所述聚乙二醇单甲醚的数均分子量为2000~10000;在某些实施例中,所述聚乙二醇单甲醚的数均分子量为4000~8000;在某些实施例中,所述聚乙二醇单甲醚的数均分子量为5000~6000。
在本发明的实施例中,所述聚乙二醇单甲醚、三乙胺和甲基磺酰氯在第二溶剂中反应完成后,还包括将得到的反应产物进行洗涤、干燥和沉降,得到中间产物。在本发明的实施例中,所述洗涤的试剂为氯化钠饱和溶液;所述洗涤的次数为4~6次;所述干燥的试剂为无水硫酸镁;所述干燥的时间为20~30h;所述沉降的试剂为乙醚或无水乙醚。在本发明的实施例中,所述沉降完成后,可以将得到的沉降产物进行真空干燥,得到中间产物。在本发明的实施例中,所述真空干燥的温度为20~30℃。在本发明的实施例中,所述真空干燥的时间为20~30h。
得到中间产物后,将所述中间产物与氨水反应,得到具有式(ⅲ)结构的端氨基聚乙二醇单甲醚。在本发明的实施例中,步骤b)中,所述反应的温度为10~40℃;在某些实施例中,所述反应的温度为15~35℃;在某些实施例中,所述反应的温度为20~30℃。在本发明的实施例中,所述中间产物和氨水反应的时间为3~7d;在某些实施例中,所述中间产物和氨水反应的时间为4~5d。在本发明的实施例中,可以在搅拌的条件下将所述中间产物和氨水进行反应。
在本发明的实施例中,所述中间产物与氨水的质量比为1:10~40。在某些实施例中,所述中间产物与氨水的质量比为1:10。
在本发明的实施例中,可以在氯化铵的存在下将中间产物和氨水进行反应,得到具有式(ⅲ)结构的端氨基聚乙二醇单甲醚。所述氯化铵起到增溶的作用。在本发明的实施例中,所述中间产物和氯化铵的质量比为1:0.7~1.5;在某些实施例中,所述中间产物和氯化铵的质量比为1:0.8~1.2;在某些实施例中,所述中间产物和氯化铵的质量比为1:0.9~1.1。
在本发明的实施例中,所述中间产物与氨水反应完成后,将得到的产物依次进行萃取、洗涤、干燥、过滤、蒸发和沉降,得到具有式(ⅲ)结构的端氨基聚乙二醇单甲醚。在本发明的实施例中,所述萃取的过程为:将所述产物与氯化钠混合,得到饱和溶液;将所述饱和溶液用二氯甲烷萃取。在本发明的实施例中,所述洗涤的试剂为饱和氯化钠溶液。在本发明的实施例中,所述洗涤的次数为2~4次。在本发明的实施例中,所述干燥的试剂为无水硫酸镁。在本发明的实施例中,所述干燥的时间为20~30h。在本发明的实施例中,所述过滤的方法为抽滤。在本发明的实施例中,所述蒸发的方法为旋蒸。在本发明的实施例中,所述沉降的试剂为乙醚或无水乙醚。在本发明的实施例中,可以将沉降后得到的产物进行真空干燥,得到具有式(ⅲ)结构的端氨基聚乙二醇单甲醚。在本发明的实施例中,所述真空干燥的温度为20~30℃。在本发明的实施例中,所述真空干燥的时间为20~30h。
在本发明的实施例中,可以先将所述具有式(ⅲ)结构的端氨基聚乙二醇单甲醚溶解于n,n-二甲基甲酰胺中,在真空和加热的条件下共沸除水。在本发明的实施例中,所述加热的温度为100~130℃;在其他的实施例中,所述加热的温度为110~120℃。本发明对所述真空的真空度并无特殊的限制,采用本领域技术人员熟知的真空度即可。在本发明的实施例中,所述除水的时间为1.5~4h;在其他的实施例中,所述除水的时间为2~3h。在本发明的实施例中,所述共沸除水采用的溶剂为n,n-二甲基甲酰胺;优选为除水后的n,n-二甲基甲酰胺。
在本发明的实施例中,所述l-缬氨酸-n-内羧酸酐具有式(ⅳ)所示的结构。所述l-缬氨酸-n-内羧酸酐的制备方法按照本领域技术人员熟知的方法制备得到,示例的,所述l-缬氨酸-n-内羧酸酐的制备方法为:
在有机溶剂中,将l-缬氨酸和三光气进行反应,得到具有式(ⅳ)所示结构的l-缬氨酸-n-内羧酸酐。
在本发明的实施例中,所述l-缬氨酸和三光气反应的温度为40~70℃;在某些实施例中,所述l-缬氨酸和三光气反应的温度为45~55℃。所述反应的时间为0.5~6h;在某些实施例中,所述反应的时间为6h。在本发明的实施例中,可以在搅拌的条件下将l-缬氨酸和三光气进行反应。在本发明的实施例中,可以在氮气的保护下将l-缬氨酸和三光气进行反应。
在本发明的实施例中,所述l-缬氨酸和三光气反应的有机溶剂为四氢呋喃。在本发明的实施例中,所述l-缬氨酸和三光气的质量比为1:0.6~1.5;在某些实施例中,所述l-缬氨酸和三光气的质量比为1:0.8~1.3;在某些实施例中,所述l-缬氨酸和三光气的质量比为1:1.5。
在本发明的实施例中,所述l-缬氨酸和三光气反应完成后,将得到的反应产物依次进行沉降、溶解、洗涤、干燥和过滤,得到l-缬氨酸-n-内羧酸酐。在本发明的实施例中,所述沉降的试剂为正己烷;所述溶解的试剂为乙酸乙酯;所述洗涤的试剂为碳酸氢钠溶液;所述干燥的试剂为无水硫酸镁。在本发明的实施例中,所述过滤的方法为抽滤;所述过滤完成后,得到具有式(ⅳ)所示结构的l-缬氨酸-n-内羧酸酐。
在本发明的实施例中,所述l-丙氨酸-n-内羧酸酐具有式(ⅴ)所示的结构。所述l-丙氨酸-n-内羧酸酐的制备方法按照本领域技术人员熟知的方法制备得到,示例的,所述l-丙氨酸-n-内羧酸酐的制备方法为:
在有机溶剂中,将丙氨酸和三光气进行反应,得到具有式(ⅴ)所示结构的l-丙氨酸-n-内羧酸酐。
在上述制备l-丙氨酸-n-内羧酸酐的过程中,所述反应的温度为50~80℃;在某些实施例中,所述反应的温度为65~75℃;在某些实施例中,所述反应的温度为50℃。所述反应的时间为2~4h;在其他的实施例中,所述反应的时间为3h。在本发明的实施例中,可以在搅拌的条件下将l-丙氨酸和三光气进行反应。在本发明的实施例中,可以在氮气的保护下将l-丙氨酸和三光气进行反应。
在上述过程中,所述有机溶剂为四氢呋喃。在本发明的实施例中,所述l-丙氨酸和三光气的质量比为1:1~2;在某些实施例中,所述l-丙氨酸和三光气的质量比为1:1.2~1.8;在某些实施例中,所述l-丙氨酸和三光气的质量比为1:1.4~1.6。
在本发明的实施例中,所述l-丙氨酸和三光气反应完成后,将得到的反应产物依次进行沉降、溶解、洗涤、干燥和过滤,得到具有式(ⅴ)所示结构的l-丙氨酸-n-内羧酸酐。所述沉降的试剂为正己烷;所述溶解的试剂为乙酸乙酯;所述洗涤的试剂为碳酸氢钠溶液;所述干燥的试剂为无水硫酸镁;所述过滤的方法为抽滤。在某些实施例中,所述抽滤后,还包括重结晶和干燥。本发明对重结晶和干燥的步骤和参数并无特殊的限制,采用本领域技术人员熟知的步骤和参数即可。
在本发明的实施例中,所述l-甘氨酸-n-内羧酸酐具有式(ⅵ)所示的结构。所述l-甘氨酸-n-内羧酸酐的制备方法按照本领域技术人员熟知的方法制备得到,示例的,所述具有式(ⅵ)所示结构的l-甘氨酸-n-内羧酸酐的制备方法为:
在有机溶剂中,将甘氨酸和三光气进行反应,得到具有式(ⅵ)所示结构的l-甘氨酸-n-内羧酸酐。
在上述制备l-甘氨酸-n-内羧酸酐的过程中,所述反应的温度为60~80℃;在某些实施例中,所述反应的温度为65~75℃;在某些实施例中,所述反应的温度为70℃。所述反应的时间为2~4h;在其他的实施例中,所述反应的时间为3h。在本发明的实施例中,可以在搅拌的条件下将l-甘氨酸和三光气进行反应。在本发明的实施例中,可以在氮气的保护下将l-丙氨酸和三光气进行反应。
在上述过程中,所述有机溶剂为四氢呋喃。在本发明的实施例中,所述l-甘氨酸和三光气的质量比为1:1~2;在某些实施例中,所述l-甘氨酸和三光气的质量比为1:1.2~1.8;在某些实施例中,所述l-甘氨酸和三光气的质量比为1:1.4~1.6。
在本发明的实施例中,所述l-甘氨酸和三光气反应完成后,将得到的反应产物依次进行沉降、溶解、洗涤、干燥和过滤,得到具有式(ⅵ)所示结构的l-甘氨酸-n-内羧酸酐。所述沉降的试剂为正己烷;所述溶解的试剂为乙酸乙酯;所述洗涤的试剂为碳酸氢钠溶液;所述干燥的试剂为无水硫酸镁;所述过滤的方法为抽滤。
在本发明的实施例中,所述l-亮氨酸-n-内羧酸酐具有式(ⅶ)所示的结构。所述l-亮氨酸-n-内羧酸酐的制备方法按照本领域技术人员熟知的方法制备得到,示例的,所述l-亮氨酸-n-内羧酸酐的制备方法为:
在有机溶剂中,将亮氨酸和三光气进行反应,得到具有式(ⅶ)所示结构的l-亮氨酸-n-内羧酸酐。
在上述制备l-亮氨酸-n-内羧酸酐的过程中,所述反应的温度为60~80℃;在某些实施例中,所述反应的温度为65~75℃;在某些实施例中,所述反应的温度为50℃。所述反应的时间为2~4h;在其他的实施例中,所述反应的时间为3h。在本发明的实施例中,可以在搅拌的条件下将l-亮氨酸和三光气进行反应。在本发明的实施例中,可以在氮气的保护下将l-丙氨酸和三光气进行反应。
在上述过程中,所述有机溶剂为四氢呋喃。在本发明的实施例中,所述l-亮氨酸和三光气的质量比为1:0.9~2;在某些实施例中,所述l-亮氨酸和三光气的质量比为1:1.2~1.8;在某些实施例中,所述l-亮氨酸和三光气的质量比为1:0.95。
在本发明的实施例中,所述l-亮氨酸和三光气反应完成后,将得到的反应产物依次进行沉降、溶解、洗涤、干燥和过滤,得到具有式(ⅶ)所示结构的l-亮氨酸-n-内羧酸酐。所述沉降的试剂为正己烷;所述溶解的试剂为乙酸乙酯;所述洗涤的试剂为碳酸氢钠溶液;所述干燥的试剂为无水硫酸镁;所述过滤的方法为抽滤。在某些实施例中,所述抽滤后,还包括重结晶和干燥。本发明对重结晶和干燥的步骤和参数并无特殊的限制,采用本领域技术人员熟知的步骤和参数即可。
本发明将具有式(ⅲ)结构的端氨基聚乙二醇单甲醚和l-氨基酸-n-内羧酸酐在第一溶剂中反应,得到具有式(ⅰ)所示结构的聚氨基酸。
本发明的实施例中,所述具有式(ⅲ)结构的端氨基聚乙二醇单甲醚和l-氨基酸-n-内羧酸酐的摩尔比为1:1~40。在某些实施例中,所述端氨基聚乙二醇单甲醚与l-缬氨酸-n-内羧酸酐的摩尔比为1:20~30或1:20.27;所述端氨基聚乙二醇单甲醚与l-丙氨酸-n-内羧酸酐的摩尔比为1:20~35或1:30.67;所述端氨基聚乙二醇单甲醚与l-甘氨酸-n-内羧酸酐的摩尔比为1:30~40或1:38;所述端氨基聚乙二醇单甲醚与l-亮氨酸-n-内羧酸酐的摩尔比为1:4~22或1:18。
本发明的实施例中,所述具有式(ⅲ)结构的端氨基聚乙二醇单甲醚和l-氨基酸-n-内羧酸酐在第一溶剂中反应的温度为4~85℃,反应的时间为60~100h。本发明的实施例中,所述反应的温度为4℃;反应的时间为72h。
本发明的实施例中,所述反应在真空的条件下进行。本发明对所述真空的真空度并无特殊的限制,采用本领域技术人员熟知的真空度即可。
本发明的实施例中,将具有式(ⅲ)结构的端氨基聚乙二醇单甲醚和l-氨基酸-n-内羧酸酐在第一溶剂中反应后的产物进行沉降和干燥,得到聚氨基酸温敏物理凝胶。在本发明的实施例中,所述沉降的试剂为乙醚或无水乙醚。在本发明的实施例中,所述沉降的次数为2~4次。
在上述第二种制备方法中,将所述具有式(ⅲ)结构的端氨基聚乙二醇单甲醚与d-氨基酸-n-内羧酸酐在第一溶剂中反应,得到具有式(ⅱ)所示结构的聚氨基酸。
具有式(ⅱ)所示结构的聚氨基酸的制备方法中,r、m和n的限定同上,在此不再赘述。式(ⅲ)中,30≤i≤200;在某些实施例中,60≤i≤190;在某些实施例中,70≤i≤180;在某些实施例中,100≤i≤150。
所述d-氨基酸-n-内羧酸酐为d-缬氨酸-n-内羧酸酐、d-丙氨酸-n-内羧酸酐、d-甘氨酸-n-内羧酸酐或d-亮氨酸-n-内羧酸酐。
在本发明的实施例中,所述d-缬氨酸-n-内羧酸酐具有式(ⅷ)所示的结构:
所述d-丙氨酸-n-内羧酸酐具有式(ⅸ)所示的结构:
所述d-甘氨酸-n-内羧酸酐具有式(ⅹ)所示的结构:
所述d-亮氨酸-n-内羧酸酐具有式(ⅺ)所示的结构:
在本发明的实施例中,所述第一溶剂为n,n-二甲基甲酰胺。
在本发明的实施例中,所述具有式(ⅲ)结构的端氨基聚乙二醇单甲醚的制备方法同上,在此不再赘述。
在本发明的实施例中,所述d-缬氨酸-n-内羧酸酐具有式(ⅷ)所示的结构。所述d-缬氨酸-n-内羧酸酐的制备方法按照本领域技术人员熟知的方法制备得到,示例的,所述d-缬氨酸-n-内羧酸酐的制备方法为:
在有机溶剂中,将d-缬氨酸和三光气进行反应,得到具有式(ⅷ)所示结构的d-缬氨酸-n-内羧酸酐。
在本发明的实施例中,所述d-缬氨酸和三光气反应的温度为40~60℃;在某些实施例中,所述d-缬氨酸和三光气反应的温度为45~55℃。在某些实施例中,所述d-缬氨酸和三光气反应的温度为50℃。所述反应的时间为0.5~4h;在某些实施例中,所述反应的时间为3h。在本发明的实施例中,可以在搅拌的条件下将d-缬氨酸和三光气进行反应。在本发明的实施例中,可以在氮气的保护下将d-缬氨酸和三光气进行反应。
在本发明的实施例中,所述d-缬氨酸和三光气反应的有机溶剂为四氢呋喃。在本发明的实施例中,所述d-缬氨酸和三光气的质量比为1:0.6~1.5;在某些实施例中,所述d-缬氨酸和三光气的质量比为1:0.8~1.3;在某些实施例中,所述d-缬氨酸和三光气的质量比为1:0.9~1.1。
在本发明的实施例中,所述d-缬氨酸和三光气反应完成后,将得到的反应产物依次进行沉降、溶解、洗涤、干燥和过滤,得到d-缬氨酸-n-内羧酸酐。在本发明的实施例中,所述沉降的试剂为正己烷;所述溶解的试剂为乙酸乙酯;所述洗涤的试剂为碳酸氢钠溶液;所述干燥的试剂为无水硫酸镁。在本发明的实施例中,所述过滤的方法为抽滤;所述过滤完成后,可以将过滤后得到的产物进行重结晶和干燥,得到具有式(ⅷ)所示结构的d-缬氨酸-n-内羧酸酐。
在本发明的实施例中,所述d-丙氨酸-n-内羧酸酐具有式(ⅸ)所示的结构。所述d-丙氨酸-n-内羧酸酐的制备方法按照本领域技术人员熟知的方法制备得到,示例的,所述d-丙氨酸-n-内羧酸酐的制备方法为:
在有机溶剂中,将d-丙氨酸和三光气进行反应,得到具有式(ⅸ)所示结构的d-丙氨酸-n-内羧酸酐。
在上述制备d-丙氨酸-n-内羧酸酐的过程中,所述反应的温度为50~80℃;在某些实施例中,所述反应的温度为65~75℃;在某些实施例中,所述反应的温度为50℃。所述反应的时间为2~4h;在某些实施例中,所述反应的时间为3h。在本发明的实施例中,可以在搅拌的条件下将d-丙氨酸和三光气进行反应。在本发明的实施例中,可以在氮气的保护下将d-丙氨酸和三光气进行反应。
在上述过程中,所述有机溶剂为四氢呋喃。在本发明的实施例中,所述d-丙氨酸和三光气的质量比为1:0.9~2;在某些实施例中,所述d-丙氨酸和三光气的质量比为1:1.2~1.8;在某些实施例中,所述d-丙氨酸和三光气的质量比为1:0.95。
在本发明的实施例中,所述d-丙氨酸和三光气反应完成后,将得到的反应产物依次进行沉降、溶解、洗涤、干燥和过滤,得到具有式(ⅸ)所示结构的d-丙氨酸-n-内羧酸酐。所述沉降的试剂为正己烷;所述溶解的试剂为乙酸乙酯;所述洗涤的试剂为碳酸氢钠溶液;所述干燥的试剂为无水硫酸镁;所述过滤的方法为抽滤。所述过滤完成后,可以将过滤后得到的产物进行重结晶和干燥。
在本发明的实施例中,所述d-甘氨酸-n-内羧酸酐具有式(ⅹ)所示的结构。所述d-甘氨酸-n-内羧酸酐的制备方法按照本领域技术人员熟知的方法制备得到,示例的,所述d-甘氨酸-n-内羧酸酐的制备方法为:
在有机溶剂中,将d-甘氨酸和三光气进行反应,得到具有式(ⅹ)所示结构的d-甘氨酸-n-内羧酸酐。
在本发明的实施例中,所述d-甘氨酸和三光气反应的温度为40~60℃;在某些实施例中,所述d-甘氨酸和三光气反应的温度为45~55℃。所述反应的时间为0.5~4h;在某些实施例中,所述反应的时间为3h。在本发明的实施例中,可以在搅拌的条件下将d-甘氨酸和三光气进行反应。在本发明的实施例中,可以在氮气的保护下将d-缬氨酸和三光气进行反应。
在本发明的实施例中,所述d-甘氨酸和三光气反应的有机溶剂为四氢呋喃。在本发明的实施例中,所述d-甘氨酸和三光气的质量比为1:0.6~1.5;在某些实施例中,所述d-甘氨酸和三光气的质量比为1:0.8~1.3;在某些实施例中,所述d-甘氨酸和三光气的质量比为1:0.9~1.1。
在本发明的实施例中,所述d-甘氨酸和三光气反应完成后,将得到的反应产物依次进行沉降、溶解、洗涤、干燥和过滤,得到d-甘氨酸-n-内羧酸酐。在本发明的实施例中,所述沉降的试剂为正己烷;所述溶解的试剂为乙酸乙酯;所述洗涤的试剂为碳酸氢钠溶液;所述干燥的试剂为无水硫酸镁。在本发明的实施例中,所述过滤的方法为抽滤;所述过滤完成后,可以将过滤后得到的产物进行重结晶和干燥,得到具有式(ⅹ)所示结构的d-甘氨酸-n-内羧酸酐。
在本发明的实施例中,所述d-亮氨酸-n-内羧酸酐具有式(ⅺ)所示的结构。所述d-亮氨酸-n-内羧酸酐的制备方法按照本领域技术人员熟知的方法制备得到,示例的,所述d-亮氨酸-n-内羧酸酐的制备方法为:
在有机溶剂中,将d-亮氨酸和三光气进行反应,得到具有式(ⅺ)所示结构的d-亮氨酸-n-内羧酸酐。
在本发明的实施例中,所述d-亮氨酸和三光气反应的温度为40~60℃;在某些实施例中,所述d-亮氨酸和三光气反应的温度为45~55℃。所述反应的时间为0.5~4h;在某些实施例中,所述反应的时间为3h。在本发明的实施例中,可以在搅拌的条件下将d-亮氨酸和三光气进行反应。在本发明的实施例中,可以在氮气的保护下将d-亮氨酸和三光气进行反应。
在本发明的实施例中,所述d-亮氨酸和三光气反应的有机溶剂为四氢呋喃。在本发明的实施例中,所述d-亮氨酸和三光气的质量比为1:0.6~1.5;在某些实施例中,所述d-亮氨酸和三光气的质量比为1:0.8~1.3;在某些实施例中,所述d-亮氨酸和三光气的质量比为1:0.9~1.1。
在本发明的实施例中,所述d-亮氨酸和三光气反应完成后,将得到的反应产物依次进行沉降、溶解、洗涤、干燥和过滤,得到d-亮氨酸-n-内羧酸酐。在本发明的实施例中,所述沉降的试剂为正己烷;所述溶解的试剂为乙酸乙酯;所述洗涤的试剂为碳酸氢钠溶液;所述干燥的试剂为无水硫酸镁。在本发明的实施例中,所述过滤的方法为抽滤;所述过滤完成后,可以将过滤后得到的产物进行重结晶和干燥,得到具有式(ⅺ)所示结构的d-亮氨酸-n-内羧酸酐。
本发明将所述具有式(ⅲ)结构的端氨基聚乙二醇单甲醚与d-氨基酸-n-内羧酸酐在第一溶剂中反应,得到具有式(ⅱ)所示结构的聚氨基酸。
本发明的实施例中,所述具有式(ⅲ)结构的端氨基聚乙二醇单甲醚与d-氨基酸-n-内羧酸酐的摩尔比为1:1~40。在某些实施例中,所述端氨基聚乙二醇单甲醚与d-缬氨酸-n-内羧酸酐的摩尔比为1:20~30或1:20.27;所述端氨基聚乙二醇单甲醚与d-丙氨酸-n-内羧酸酐的摩尔比为1:20~28或1:27.07;所述端氨基聚乙二醇单甲醚与d-甘氨酸-n-内羧酸酐的摩尔比为1:30~40或1:33.8;所述端氨基聚乙二醇单甲醚与d-亮氨酸-n-内羧酸酐的摩尔比为1:5~20或1:13.8。
本发明的实施例中,所述具有式(ⅲ)结构的端氨基聚乙二醇单甲醚和d-氨基酸-n-内羧酸酐在第一溶剂中反应的温度为4~85℃,反应的时间为60~100h。本发明的实施例中,所述反应的温度为4℃;反应的时间为72h。
本发明的实施例中,所述反应在真空的条件下进行。本发明对所述真空的真空度并无特殊的限制,采用本领域技术人员熟知的真空度即可。
本发明的实施例中,将具有式(ⅲ)结构的端氨基聚乙二醇单甲醚和d-氨基酸-n-内羧酸酐在第一溶剂中反应后的产物进行沉降和干燥,得到聚氨基酸温敏物理凝胶。在本发明的实施例中,所述沉降的试剂为乙醚或无水乙醚。在本发明的实施例中,所述沉降的次数为2~4次。
本发明还提供了一种聚氨基酸水凝胶,由上文所述的聚氨基酸和溶剂组成;或由上文所述制备方法制得的聚氨基酸和溶剂组成。在本发明的实施例中,所述溶剂可以为磷酸缓冲盐溶液(pbs缓冲液)或水。在某些实施例中,所述pbs缓冲液由1.42gna2hpo4、0.27gkh2po4、8gnacl和0.2gkcl溶解在1l水中得到。
所述聚氨基酸水凝胶按照本领域技术人员熟知的方式制备即可,示例的,所述聚氨基酸水凝胶的制备方法为:
将聚氨基酸进行溶解,升高温度,得到聚氨基酸水凝胶。
本发明的实施例中,制备聚氨基酸水凝胶的过程中,所述溶解的温度为0~10℃;在某些实施例中,所述溶解的温度为5~15℃;在某些实施例中,所述溶解的温度为10~20℃。在本发明的实施例中,所述溶解的时间为12~24h;在某些实施例中,所述溶解的时间为14~23h;在某些实施例中,所述溶解的时间为16~22h。在本发明的实施例中,可以在搅拌的条件下进行所述溶解。在本发明的实施例中,所述聚氨基酸溶解的溶剂可以为磷酸缓冲盐溶液(pbs)或水。
本发明的实施例中,所述聚氨基酸溶解后,逐渐升高温度,得到水凝胶。在本发明的实施例中,所述水可以为蒸馏水;在某些实施例中,所述水可以为二次蒸馏水。在本发明的实施例中,所述溶解后的溶液质量浓度为1~20%;在某些实施例中,所述溶解后的溶液质量浓度为2~10%;在某些实施例中,所述溶解后的溶液质量浓度为5~20%。
本发明提供的不同构型的聚氨基酸,经历溶胶-凝胶转化过后,皮下注射,观察所引起的局部炎症,可以发现,得到的聚氨基酸水凝胶在体内能够引起较大的炎症反应。炎症反应可以通过产生一系列炎症因子刺激t细胞、dc细胞等产生一系列免疫响应,从而达到免疫治疗的效果。另外,本发明的聚氨基酸的成胶性能可通过温度、ph、光与电等实现可调控性,因此,本申请提供的水凝胶具有温度响应性。
本发明还提供了一种载药凝胶,包括聚氨基酸和负载在聚氨基酸上的抗肿瘤药物,所述聚氨基酸为上文所述的聚氨基酸或上文所述制备方法制得的聚氨基酸。在本发明中,所述聚氨基酸在低温下能够自发组装成具有核-壳结构的纳米尺寸的凝胶,可以负载抗肿瘤药物形成载药凝胶,温度升高形成可降解的载药凝胶,本发明提供的载药凝胶通过增强渗透和长的代谢时间使其中的抗肿瘤药物在肿瘤部位实现缓慢长效治疗。
在本发明中,所述抗肿瘤药物为紫杉醇。本发明对所述聚氨基酸和抗肿瘤药物的用量没有特殊的限制,采用不同用量的聚氨基酸和抗肿瘤药物可以得到不同载药率的载药凝胶。
所述载药凝胶按照本领域技术人员熟知的方式制备即可,示例的,所述载药凝胶的制备方法为:
将聚氨基酸和抗肿瘤药物在有机溶剂中混合,得到载药凝胶。
在上述制备载药凝胶的过程中,所述混合的温度为0~25℃;在某些实施例中,所述混合的温度为10~25℃;在某些实施例中,所述混合的温度为25℃。在本发明的实施例中,所述混合的时间为10~30h;在某些实施例中,所述混合的时间为10~20h;在某些实施例中,所述混合的时间为20h。在本发明的实施例中,可以在搅拌的条件下进行所述混合。在本发明的实施例中,所述聚氨基酸和抗肿瘤药物混合的有机溶剂可以为n,n-二甲基甲酰胺(dmf)。
本发明的实施例中,所述聚氨基酸和抗肿瘤药物混合后,将得到的混合物与水混合后进行透析,得到载药凝胶。在本发明的实施例中,所述水可以为蒸馏水;在某些实施例中,所述水可以为二次蒸馏水。在本发明的实施例中,所述水的用量与将聚氨基酸和抗肿瘤药物混合时的有机溶剂体积一致。在本发明的实施例中,可以将水滴加到所述混合物中进行混合。在本发明的实施例中,所述水的滴加速度为0.1~1ml/min;在某些实施例中,所述水的滴加速度为0.2~0.8ml/min;在某些实施例中,所述水的滴加速度为0.5ml/min。
在本发明的实施例中,所述透析过程中的截留分子量为3000~4000dalton;在某些实施例中,所述透析过程中的截留分子量为3200~3800dalton;在某些实施例中,所述透析过程中的截留分子量为3500dalton。在本发明的实施例中,所述透析的时间为8~16h;在某些实施例中,所述透析的时间为9~15h;在某些实施例中,所述透析的时间为12h。在本发明的实施例中,可以采用透析袋进行透析。
本发明对上述所采用的原料的来源并无特殊的限制,可以为一般市售。
本发明提供了一种聚氨基酸,具有式(ⅰ)或式(ⅱ)所示结构,其中,r为-h、-ch3、(ch3)2ch-或ch3)2chch2-;30≤m≤200,4≤n≤50。本申请提供的聚氨基酸一端是亲水的聚乙二醇单甲醚,另一端分别是疏水的聚-l-丙氨酸、聚-l-缬氨酸、聚-l-甘氨酸、聚-l-亮氨酸、聚-d-丙氨酸、聚-d-缬氨酸、聚-d-甘氨酸或聚-d-亮氨酸,该种具有亲水链段和疏水链段的两亲性聚氨基酸在水中能够形成纳米尺寸的物理凝胶,且在一定温度下能够成胶,因此该种物理凝胶能够作为药物传输和控制释放的载体材料。将本发明提供的聚氨基酸的水凝胶经皮下注射后,会引起较大的炎症反应,炎症反应可以通过产生一系列炎症因子刺激t细胞、dc细胞等产生一系列免疫响应,从而达到免疫治疗的效果。另外,本发明的聚氨基酸的成胶性能可通过温度、ph、光与电等实现可调控性,因此,本申请提供的水凝胶具有温度响应性。此外,本申请提供的载药凝胶中含有聚氨基酸,使该种载药凝胶具有较好的生物相容性、低毒性和生物降解性。
为了进一步说明本发明,下面结合实施例对本发明提供的一种聚氨基酸、其制备方法及应用进行详细地描述,但不能将它们理解为对本发明保护范围的限定。
以下实施例所用的原料均为市售商品。
实施例1
将20g(0.01mol)的数均分子量为2000的聚乙二醇单甲醚放入干燥的反应瓶中,然后在120℃且搅拌的条件下抽真空2h,将反应瓶进行油浴,当温度为60℃时,分别向反应瓶中加入10g的无水硫酸镁,200ml除过水的二氯甲烷,在冰水浴的条件向反应瓶中加入5ml(0.036mol)的三乙胺,30ml(0.39mol)的甲基磺酰氯,在25℃、搅拌子搅拌条件下反应72h,反应结束后,将得到的反应产物用饱和的氯化钠溶液洗涤五次后加入无水硫酸镁干燥过夜;将干燥后的产物用无水乙醚沉降,在25℃下真空干燥24h后,得到中间产物;
将所述中间产物和与所述中间产物等质量的氯化铵溶于氨水中,氨水的质量为中间产物质量的10倍,在25℃搅拌条件下反应4d,将得到的反应产物用二氯甲烷萃取,将萃取后的产物用饱和氯化钠溶液洗涤三次后加入无水硫酸镁干燥12h,抽滤,将液体旋蒸至150ml,将旋蒸后的产物用无水乙醚沉降两次,在25℃下真空干燥24h后,得到端氨基聚乙二醇单甲醚。
对本发明实施例1制备得到的端氨基聚乙二醇单甲醚进行核磁共振检测,结果如图1所示。图1为本发明实施例1得到的端氨基聚乙二醇单甲醚的13c核磁谱图。检测结果为,本发明实施例1制备得到的端氨基聚乙二醇单甲醚具有式(1)所示的结构:
式(1)中,n为45。
按照下述公式计算本发明实施例1制备得到的端氨基聚乙二醇单甲醚的产率:
产率=实际得到的产物质量/理论得到的产物质量×100%;
计算结果为,本发明实施例1制备得到的端氨基聚乙二醇单甲醚的产率为89.8%。
实施例2
将20g(0.005mol)的数均分子量为4000的聚乙二醇放入干燥的反应瓶中,然后在120℃且搅拌的条件下抽真空2h,将反应瓶进行油浴,当温度为60℃时,分别向反应瓶中加入10g的无水硫酸镁,200ml除过水的二氯甲烷,在冰水浴的条件向反应瓶中加入1.25ml(0.0089mol)的三乙胺,7.5ml(0.097mol)的甲基磺酰氯,在25℃、搅拌子搅拌条件下反应72h,反应结束后,将得到的反应产物用饱和的氯化钠溶液洗涤五次后加入无水硫酸镁干燥过夜;将干燥后的产物用无水乙醚沉降,在25℃下真空干燥24h后,得到中间产物;
将所述中间产物和与所述中间产物等质量的氯化铵溶于氨水中,氨水的质量为中间产物质量的10倍,在25℃搅拌条件下反应4d,将得到的反应产物用二氯甲烷萃取,将萃取后的产物用饱和氯化钠溶液洗涤三次后加入无水硫酸镁干燥12h,抽滤,将液体旋蒸至150ml,将旋蒸后的产物用无水乙醚沉降两次,在25℃下真空干燥24h后,得到端氨基聚乙二醇。
对本发明实施例2制备得到的端氨基聚乙二醇进行核磁共振检测,检测结果为,本发明实施例2制备得到的端氨基聚乙二醇具有式(2)所示的结构:
式(2)中,n为90。
按照实施例1所述的方法计算本发明实施例2制备得到的端氨基聚乙二醇的产率;结果为,本发明实施例2制备得到的端氨基聚乙二醇的产率为83.5%。
实施例3
将20g(0.004mol)的数均分子量为5000的聚乙二醇单甲醚放入干燥的反应瓶中,然后在120℃且搅拌的条件下抽真空2h,将反应瓶进行油浴,当温度为60℃时,分别向反应瓶中加入10g的无水硫酸镁,200ml除过水的二氯甲烷,在冰水浴的条件向反应瓶中加入2ml(0.014mol)的三乙胺,12ml(0.155mol)的甲基磺酰氯,在25℃、搅拌子搅拌条件下反应72h,反应结束后,将得到的反应产物用饱和的氯化钠溶液洗涤五次后加入无水硫酸镁干燥过夜;将干燥后的产物用无水乙醚沉降,在25℃下真空干燥24h后,得到中间产物;
将所述中间产物和与所述中间产物等质量的氯化铵溶于氨水中,氨水的体积为中间产物质量的10倍,在25℃搅拌条件下反应4d,将得到的反应产物用二氯甲烷萃取,将萃取后的产物用饱和氯化钠溶液洗涤三次后加入无水硫酸镁干燥12h,抽滤,将液体旋蒸至150ml,将旋蒸后的产物用无水乙醚沉降两次,在25℃下真空干燥24h后,得到端氨基聚乙二醇单甲醚。
对本发明实施例3制备得到的端氨基聚乙二醇单甲醚进行核磁共振检测,检测结果如图1和图2所示,图1为本发明实施例3制备得到的端氨基聚乙二醇单甲醚核磁共振谱图,图2为本发明实施例3制备得到的端氨基聚乙二醇单甲醚红外谱图,由图1和图2可知,本发明实施例3制备得到的端氨基聚乙二醇单甲醚具有式(3)所示的结构:
式(3)中,n为114。
按照实施例1所述的方法计算本发明实施例3制备得到的端氨基聚乙二醇单甲醚的产率;计算结果为,本发明实施例3制备得到的端氨基聚乙二醇单甲醚的产率为95.9%。
实施例4
在70℃加热搅拌的条件下,用除过水的四氢呋喃将10g的l-缬氨酸溶解于干燥的三口瓶中,通氮气,加入6g的三光气,1h后再加入5g的三光气,2h后再加入4g的三光气,共搅拌回流3h;将得到的反应产物用氮气吹半个小时后,依次用冷的正己烷沉降,冷的乙酸乙酯溶解,冷的碳酸氢钠溶液洗涤一次,再用冷水洗涤三次,将洗涤后的产物和无水硫酸镁混合后放入-20℃冰箱中干燥过夜;将干燥后的产物进行抽滤,将溶液中的溶剂抽干后,得到具有式(ⅳ)所示结构的l-缬氨酸-n-内羧酸酐。
实施例5
在50℃加热搅拌条件下,用除过水的四氢呋喃将20g的l-丙氨酸溶解于干燥的三口瓶中,通氮气,加入12g的三光气,1h后再加入5g的三光气,2h后再加入2g的三光气2g,搅拌至澄清;将得到的反应产物用氮气吹半个小时后,依次用冷的正己烷沉降,冷的乙酸乙酯溶解,冷的碳酸氢钠溶液洗涤一次,再用冷水洗涤三次,将洗涤后的产物和无水硫酸镁混合后放入冰箱中干燥过夜;将干燥后的产物抽滤,将溶液中的溶剂抽干后重结晶、干燥,得到具有式(ⅴ)所示结构的l-丙氨酸-n-内羧酸酐。
实施例6
在70℃加热搅拌的条件下,用除过水的四氢呋喃将10g的l-甘氨酸溶解于干燥的三口瓶中,通氮气,加入6g的三光气,一个小时后再加入5g的三光气,两个小时后再加入4g的三光气,共搅拌回流3个小时;将得到的反应产物用氮气吹半个小时后,依次用冷的正己烷沉降,冷的乙酸乙酯溶解,冷的碳酸氢钠溶液洗涤一次,再用冷水洗涤三次,将洗涤后的产物和无水硫酸镁混合后放入-20℃冰箱中干燥过夜;将干燥后的产物进行抽滤,将溶液中的溶剂抽干后,得到具有式(ⅵ)所示结构的l-甘氨酸-n-内羧酸酐。
实施例7
在50℃加热搅拌条件下,用除过水的四氢呋喃将20g的l-亮氨酸溶解于干燥的三口瓶中,通氮气,加入12g的三光气,1h后再加入5g的三光气,2h后再加入2g的三光气,搅拌至澄清;将得到的反应产物用氮气吹半个小时后,依次用冷的正己烷沉降,冷的乙酸乙酯溶解,冷的碳酸氢钠溶液洗涤一次,再用冷水洗涤三次,将洗涤后的产物和无水硫酸镁混合后放入冰箱中干燥过夜;将干燥后的产物抽滤,将溶液中的溶剂抽干后重结晶、干燥,得到具有式(ⅶ)所示结构的l-亮氨酸-n-内羧酸酐。
实施例8
在50℃加热搅拌条件下,用除过水的四氢呋喃将20g的d-缬氨酸溶解于干燥的三口瓶中,通氮气,加入12g的三光气,1h后再加入5g的三光气,2h后再加入2g的三光气,搅拌至澄清;将得到的反应产物用氮气吹半个小时后,依次用冷的正己烷沉降,冷的乙酸乙酯溶解,冷的碳酸氢钠溶液洗涤一次,再用冷水洗涤三次,将洗涤后的产物和无水硫酸镁混合后放入冰箱中干燥过夜;将干燥后的产物抽滤,将溶液中的溶剂抽干后重结晶、干燥,得到具有式(ⅷ)所示结构的d-缬氨酸-n-内羧酸酐。
实施例9
在50℃加热搅拌条件下,用除过水的四氢呋喃将20g的d-丙氨酸溶解于干燥的三口瓶中,通氮气,加入12g的三光气,1h后再加入5g的三光气,2h后再加入2g的三光气,搅拌至澄清;将得到的反应产物用氮气吹半个小时后,依次用冷的正己烷沉降,冷的乙酸乙酯溶解,冷的碳酸氢钠溶液洗涤一次,再用冷水洗涤三次,将洗涤后的产物和无水硫酸镁混合后放入冰箱中干燥过夜;将干燥后的产物抽滤,将溶液中的溶剂抽干后重结晶、干燥,得到具有式(ⅸ)所示结构的d-丙氨酸-n-内羧酸酐。
实施例10
在50℃加热搅拌条件下,用除过水的四氢呋喃将20g的d-甘氨酸溶解于干燥的三口瓶中,通氮气,加入12g的三光气,1h后再加入5g的三光气,2h后再加入2g的三光气,搅拌至澄清;将得到的反应产物用氮气吹半个小时后,依次用冷的正己烷沉降,冷的乙酸乙酯溶解,冷的碳酸氢钠溶液洗涤一次,再用冷水洗涤三次,将洗涤后的产物和无水硫酸镁混合后放入冰箱中干燥过夜;将干燥后的产物抽滤,将溶液中的溶剂抽干后重结晶、干燥,得到具有式(ⅹ)所示结构的d-甘氨酸-n-内羧酸酐。
实施例11
在50℃加热搅拌条件下,用除过水的四氢呋喃将20g的d-亮氨酸溶解于干燥的三口瓶中,通氮气,加入12g的三光气,1h后再加入5g的三光气,2h后再加入2g的三光气,搅拌至澄清;将得到的反应产物用氮气吹半个小时后,依次用冷的正己烷沉降,冷的乙酸乙酯溶解,冷的碳酸氢钠溶液洗涤一次,再用冷水洗涤三次,将洗涤后的产物和无水硫酸镁混合后放入冰箱中干燥过夜;将干燥后的产物抽滤,将溶液中的溶剂抽干后重结晶、干燥,得到具有式(ⅺ)所示结构的d-亮氨酸-n-内羧酸酐。
实施例12
将3g(0.0015mol)实施例1制备得到的端氨基聚乙二醇单甲醚用100ml甲苯溶解,在抽真空和120℃加热的条件下进行共沸除水,共沸除水2h后,加入除过水的200ml的n,n-二甲基甲酰胺进行溶解,向得到的溶液中加入4.32g(0.0304mol)的实施例4制备得到的l-缬氨酸-n-内羧酸酐,在抽真空0.5h后4℃下反应72h;将得到的反应产物用无水乙醚沉降三次,干燥后得到聚氨基酸。对本发明实施例12制备得到的聚氨基酸进行核磁共振检测,结果如图2所示。图2为本发明实施例12制备得到的聚氨基酸的1h核磁谱图。结果表明,本实施例制备得到的聚氨基酸为具有式(4)所示结构的聚-l-缬氨酸。
式(4)中,m为45,n为17。按照实施例1所述的方法计算本发明实施例的聚氨基酸的产率为84.5%。
实施例13
将3g(0.0015mol)实施例1制备得到的端氨基聚乙二醇单甲醚用100ml甲苯溶解,在抽真空和120℃加热的条件下进行共沸除水,共沸除水2h后,加入除过水的200ml的n,n-二甲基甲酰胺进行溶解,向得到的溶液中加入4.92g(0.046mol)的实施例5制备得到的l-丙氨酸-n-内羧酸酐,在抽真空0.5h后4℃下反应72h;将得到的反应产物用无水乙醚沉降三次,干燥后得到聚氨基酸。对本发明实施例13制备得到的聚氨基酸进行核磁共振检测,结果如图3所示。图3为本发明实施例13制备得到的聚氨基酸的1h核磁谱图。结果表明,本实施例制备得到的聚氨基酸为具有式(5)所示结构的聚-l-丙氨酸。
式(5)中,m为45,n为27。按照实施例1所述的方法计算本发明实施例的聚氨基酸的产率为80%。
实施例14
将3g(0.0015mol)实施例1制备得到的端氨基聚乙二醇单甲醚用100ml甲苯溶解,在抽真空和120℃加热的条件下进行共沸除水,共沸除水2h后,加入除过水的200ml的n,n-二甲基甲酰胺进行溶解,向得到的溶液中加入5.27g(0.057mol)的实施例6制备得到的l-甘氨酸-n-内羧酸酐,在抽真空0.5h后4℃下反应72h;将得到的反应产物用无水乙醚沉降三次,干燥后得到具有式(6)所示结构的聚-l-甘氨酸。
式(6)中,m为45,n为33。按照实施例1所述的方法计算本发明实施例的聚氨基酸的产率为85.7%。
实施例15
将3g(0.0015mol)实施例1制备得到的端氨基聚乙二醇单甲醚用100ml甲苯溶解,在抽真空和120℃加热的条件下进行共沸除水,共沸除水2h后,加入除过水的200ml的n,n-二甲基甲酰胺进行溶解,向得到的溶液中加入4.15g(0.027mol)的实施例7制备得到的l-亮氨酸-n-内羧酸酐,在抽真空0.5h后4℃下反应72h;将得到的反应产物用无水乙醚沉降三次,干燥后得到具有式(7)所示结构的聚-l-亮氨酸。
式(7)中,m为45,n为15。按照实施例1所述的方法计算本发明实施例的聚氨基酸的产率为82.3%。
实施例16
将3g(0.0015mol)实施例1制备得到的端氨基聚乙二醇单甲醚用100ml甲苯溶解,在抽真空和120℃加热的条件下进行共沸除水,共沸除水2h后,加入除过水的200ml的n,n-二甲基甲酰胺进行溶解,向得到的溶液中加入4.32g(0.0304mol)的实施例8制备得到的d-缬氨酸-n-内羧酸酐,在抽真空0.5h后4℃下反应72h;将得到的反应产物用无水乙醚沉降三次,干燥后得到聚氨基酸。对本发明实施例16制备得到的聚氨基酸进行核磁共振检测,结果如图4所示。图4为本发明实施例16制备得到的聚氨基酸的1h核磁谱图。结果表明,本实施例制备得到的聚氨基酸为具有式(8)所示结构的聚-d-缬氨酸。
式(8)中,m为45,n为17。按照实施例1所述的方法计算本发明实施例的聚氨基酸的产率为80.7%。
实施例17
将3g(0.0015mol)实施例1制备得到的端氨基聚乙二醇单甲醚用100ml甲苯溶解,在抽真空和120℃加热的条件下进行共沸除水,共沸除水2h后,加入除过水的200ml的n,n-二甲基甲酰胺进行溶解,向得到的溶液中加入4.92g(0.0406mol)的实施例11制备得到的d-丙氨酸-n-内羧酸酐,在抽真空0.5h后4℃下反应72h;将得到的反应产物用无水乙醚沉降三次,干燥后得到聚氨基酸。对本发明实施例17制备得到的聚氨基酸进行核磁共振检测,结果如图5所示。图5为本发明实施例17制备得到的聚氨基酸的1h核磁谱图。结果表明,本实施例制备得到的聚氨基酸为具有式(9)所示结构的聚-d-丙氨酸。
式(9)中,m为45,n为27。按照实施例1所述的方法计算本发明实施例的聚氨基酸的产率为83.6%。
实施例18
将3g(0.0015mol)实施例1制备得到的端氨基聚乙二醇单甲醚用100ml甲苯溶解,在抽真空和120℃加热的条件下进行共沸除水,共沸除水2h后,加入除过水的200ml的n,n-二甲基甲酰胺进行溶解,向得到的溶液中加入5.27g(0.0507mol)的实施例10制备得到的d-甘氨酸-n-内羧酸酐,在抽真空0.5h后4℃下反应72h;将得到的反应产物用无水乙醚沉降三次,干燥后得到具有式(10)所示结构的聚-d-甘氨酸。
式(10)中,m为45,n为32。按照实施例1所述的方法计算本发明实施例的聚氨基酸的产率为79.8%。
实施例19
将3g(0.0015mol)实施例1制备得到的端氨基聚乙二醇单甲醚用100ml甲苯溶解,在抽真空和120℃加热的条件下进行共沸除水,共沸除水2h后,加入除过水的200ml的n,n-二甲基甲酰胺进行溶解,向得到的溶液中加入4.15g(0.0207mol)的实施例11制备得到的d-亮氨酸-n-内羧酸酐,在抽真空0.5h后4℃下反应72h;将得到的反应产物用无水乙醚沉降三次,干燥后得到具有式(11)所示结构的聚-d-亮氨酸。
式(11)中,m为45,n为15。按照实施例1所述的方法计算本发明实施例的聚氨基酸的产率为86.2%。
实施例20
将实施例13得到的聚-l-丙氨酸配制成不同浓度的聚合物溶液,在0℃充分溶解。然后将聚合物溶液0.2ml转移到小瓶中,放置在水浴中。整个过程从5℃开始,每10min升高2℃,并通过倒置法观察是否成胶,如果聚合物溶液在30s内不发生流动,则认为其发生了凝胶转变,直至65℃结束。随着温度升高,发生转变的凝胶在倒置时发生流动则表明是溶胶现象。每个样品平行测试二次。如图6所示。图6为本发明实施例20得到的凝胶的相图。从图6可以看出,5~30℃之间,发生的是凝胶现象,30~65℃之间,发生的是溶胶现象。可选择较为合适的成胶浓度来进行体内实验,以符合哺乳动物的37℃的体温,因此,选择40mg/ml的浓度作为体内实验的浓度。
实施例21
将实施例17得到的聚-d-丙氨酸配制成不同浓度的聚合物溶液,在0℃充分溶解。然后将聚合物溶液0.2ml转移到小瓶中,放置在水浴中。整个过程从5℃开始,每10min升高2℃,并通过倒置法观察是否成胶,如果聚合物溶液在30s内不发生流动,则认为其发生了凝胶转变,直至65℃结束。随着温度升高,发生转变的凝胶在倒置时发生流动则表明是溶胶现象。每个样品平行测试二次。如图7所示。图7为本发明实施例21得到的凝胶的相图。从图7可以看出,5~35℃之间,发生的是凝胶现象,35~65℃之间,发生的是溶胶现象。可选择较为合适的成胶浓度来进行体内实验,以符合哺乳动物的37℃的体温,因此,选择40mg/ml的浓度作为体内实验的浓度。
实施例22
将实施例13和17得到的聚合物分别配置成浓度为0.1mg/ml的溶液,通过圆二色谱仪测其结构,如图8所示,不同构型的材料得到了关于横坐标轴对称的特征峰。图8为本发明实施例13所得的聚-l-丙氨酸与实施例17所得的聚-d-丙氨酸的圆二色谱图,由圆二色谱可知,两种材料具有相反的特征峰,表明两种材料具有不同的手性。
实施例23
将40mg本发明实施例12制备得到的聚-l-缬氨酸溶于1ml的去离子水中,待完全溶解后,吸取0.5ml打入大鼠皮下,三天后取凝胶处组织,进行苏木精-伊红染色观察其炎症反应,结果如图9所示。图9为本发明实施例23得到的凝胶处组织的苏木精-伊红染色效果图。从图9可以看出,聚-l-缬氨酸所引起的炎症较轻,因而,呈现出炎症现象。
实施例24
将40mg本发明实施例13制备得到的聚-l-丙氨酸溶于1ml的去离子水中,待完全溶解后,吸取0.5ml打入大鼠皮下,三天后取凝胶处组织,进行苏木精-伊红染色观察其炎症反应,结果如图10所示。图10为本发明实施例24得到的凝胶处组织的苏木精-伊红染色效果图。从图10可以看出,聚-l-丙氨酸引起的炎症较轻。
实施例25
将40mg本发明实施例14制备得到的聚-l-甘氨酸溶于1ml的去离子水中,待完全溶解后,吸取0.5ml打入大鼠皮下,三天后取凝胶处组织,进行苏木精-伊红染色观察其炎症反应,可观察到炎症现象。
实施例26
将40mg本发明实施例15制备得到的聚-l-亮氨酸溶于1ml的去离子水中,待完全溶解后,吸取0.5ml打入大鼠皮下,三天后取凝胶处组织,进行苏木精-伊红染色观察其炎症反应,可观察到炎症现象。
实施例27
将40mg本发明实施例16制备得到的聚-d-缬氨酸溶于1ml的去离子水中,待完全溶解后,吸取0.5ml打入大鼠皮下,三天后取凝胶处组织,进行苏木精-伊红染色观察其炎症反应,结果如图11所示。图11为本发明实施例27得到的凝胶处组织的苏木精-伊红染色效果图。从图11可以看出,聚-d-缬氨酸引起了较重的炎症现象。
实施例28
将40mg本发明实施例17制备得到的聚-d-丙氨酸溶于1ml的去离子水中,待完全溶解后,吸取0.5ml打入大鼠皮下,三天后取凝胶处组织,进行苏木精-伊红染色观察其炎症反应,结果如图12所示。图12为本发明实施例28得到的凝胶处组织的苏木精-伊红染色效果图。从图12可以看出,聚-d-丙氨酸在体内引起较重的炎症现象。
实施例29
将40mg本发明实施例18制备得到的聚-d-甘氨酸溶于1ml的去离子水中,待完全溶解后,吸取0.5ml打入大鼠皮下,三天后取凝胶处组织,进行苏木精-伊红染色观察其炎症反应,可观察到较重的炎症现象。
实施例30
将40mg本发明实施例19制备得到的聚-d-亮氨酸溶于1ml的去离子水中,待完全溶解后,吸取0.5ml打入大鼠皮下,三天后取凝胶处组织,进行苏木精-伊红染色观察其炎症反应,可观察到较重的炎症现象。
实施例31
将20mg本发明实施例12制备得到的聚氨基酸溶于3ml的dmf中,得到第一溶液;称取2mg的紫杉醇溶于2ml的dmf中,待完全溶解后,得到第二溶液;将第一溶液和第二溶液混合,在25℃下搅拌20h,向得到的混合液中以0.5ml/min的速度滴加5ml的二次蒸馏水,在25℃条件下搅拌18h后用截留分子量为3500dalton的透析袋透进行12h的透析,得到载药凝胶。
按照下述方法,计算本发明实施例28制备得到的载药凝胶的实际载药率、理论载药率和载药效率:将载药凝胶溶于n,n-二甲基甲酰胺中,载药凝胶的浓度为0.05~0.2mg/l;将紫杉醇溶于n,n-二甲基甲酰胺中,紫杉醇的浓度为0.2mg/l,将紫杉醇溶液逐级稀释;使用荧光分度计测量测试载药胶束溶液中的紫杉醇含量,得到相应的载药凝胶的实际载药效率。本实施例中的紫杉醇可以替换为阿霉素。理论载药率计算方法为:
理论载药率(wt%)=投入药物总量/(药物总量+凝胶的量)×100;
载药效率为的计算方法为:
载药效率(wt%)=实际载入凝胶中药物的量/加入药物的量×100;
测试结果为,本发明实施例28制备得到的载药凝胶的实际载药率为8.05%,理论载药率为9.09%,载药效率为88.1%。
将上述聚氨基酸分别替换为实施例13~19的聚氨基酸,按照同样的检测方法,得出:实施例13制备得到的载药凝胶的实际载药率为8%,理论载药率为9.12%,载药效率为79.6%;实施例14制备得到的载药凝胶的实际载药率为9.23%,理论载药率为9.431%,载药效率为84.4%;实施例15制备得到的载药凝胶的实际载药率为8.12%,理论载药率为9.33%,载药效率为81.4%;实施例16制备得到的载药凝胶的实际载药率为7.96%,理论载药率为8.99%,载药效率为79.8%;实施例17制备得到的载药凝胶的实际载药率为8.04%,理论载药率为8.82%,载药效率为89.3%;实施例18制备得到的载药凝胶的实际载药率为7.66%,理论载药率为8.44%,载药效率为81.1%;实施例19制备得到的载药凝胶的实际载药率为7.94%,理论载药率为8.85%,载药效率为77.6%。
对所公开的实施例的上述说明,使本领域专业技术人员能够实现或使用本发明。对这些实施例的多种修改对本领域的专业技术人员来说将是显而易见的,本文中所定义的一般原理可以在不脱离本发明的精神或范围的情况下,在其它实施例中实现。因此,本发明将不会被限制于本文所示的这些实施例,而是要符合与本文所公开的原理和新颖特点相一致的最宽的范围。
- 还没有人留言评论。精彩留言会获得点赞!