苯并噁二唑类化合物及其制备方法和医药用途与流程
本发明属于生物医药领域,具体涉及一种具有pd-1/pd-l1抑制活性的苯并噁二唑类化合物,本发明还涉及该类化合物的制备方法及其作为pd-1/pd-l1抑制剂的医药用途及用于肿瘤免疫治疗的药物组合物。
背景技术:
程序性死亡受体1(pd-1)主要表达于t细胞表面,是机体免疫检查点通路中的重要因子。而肿瘤细胞表面高表达pd-1的配体,即程序性死亡配体1(pd-l1),当其与t细胞表面的pd-1结合后,pd-1的itim(tyr223)和itsm(tyr248)酪氨酸基序分别被磷酸化,随后募集蛋白酪氨酸磷酸酶shp-2和shp-1,通过去磷酸化信号传导中间体(例如cd3ζ和plcγ1等),导致t细胞受体(tcr)信号的下调,使肿瘤细胞逃脱机体的免疫监控(immunity2016,44(5):955-972)。pd-1与pd-l1结合后还可以通过减少与效应细胞功能相关的细胞因子(如ifn-γ、il-2和tnfα)和转录因子(如gata-3、t-bet和eomes)的表达来抑制t细胞的免疫功能(sciencesignaling2012,5(230):ra46)。研究表明,抑制pd-1/pd-l1介导的免疫检查点通路,可以阻断肿瘤对t细胞的免疫抑制作用,从而激活t细胞对肿瘤的杀伤作用。
与传统的抗肿瘤疗法相比,免疫疗法最大的优势之一,就是疗效具有持久性。比如,在黑色素瘤中,欧美20%左右的晚期患者能实现临床治愈。相对于靶向疗法,免疫疗法具有更广谱的抗癌效果,而相对于化疗,它的整体副作用要小的多。目前上市的pd-1/pd-l1抑制剂都是单克隆抗体类抑制剂,有bms公司的nivolumab、merck公司的pembrolizumab、罗氏的atezolizumab和阿斯利康的durvaluman。而在研的pd-1/pd-l1抗体抑制剂更是多达几十种。目前pd-1/pd-l1小分子抑制剂还处于前期研发阶段。curis和aurigene公司联合开发的ca-170目前已完成i期临床研究(wo2016142833)。bms公司公开了一类苄基苯基醚类小分子pd1/pd-l1抑制剂(wo2015160641;wo2015034820)。小分子抑制剂相对于生物大分子具有明显的优势,由于其分子量较小,透膜性强,可以对一些实体瘤产生更好的疗效。并且小分子药物往往具有更好的生物利用度和依从性,具有更好的药代动力学参数,适合口服给药,且成本较低。
综上所述,临床上亟需开发活性高、毒副作用小的新型pd-1/pd-l1小分子抑制剂。
技术实现要素:
发明目的:针对现有技术存在的问题,本发明提供一种新型苯并噁二唑类化合物,本发明的苯并噁二唑类化合物对pd-1/pd-l1蛋白-蛋白相互作用具有明显的抑制作用,因而可应用在制备具有pd-1/pd-l1抑制活性的抑制剂,并作为免疫检查点抑制剂应用于肿瘤的免疫治疗。
本发明还提供所述苯并噁二唑类化合物及其中间体的制备方法、药物组合物及其医药用途。
技术方案:为了实现上述目的,本发明提供一种如下式(i)所示的苯并噁二唑类化合物:
r1选自:h、c1-c4烷基、x取代的c1-c4烷基、杂环烷基或-(ch2)nar;所述x选自f、cl、br、i、oh、c(o)oh、c(o)nh2、nh2、吗啉基、哌啶基、哌嗪基、四氢吡咯基或n,n-二甲基氨基;所述n是1、2、3或4;ar是取代或非取代的芳基或杂芳基;
r2选自:-(ch2)mcho、-(ch2)moh或-(ch2)mnr8r9;
m是0、1、2、3或4;
r8选自:h、c1-c4烷基或苄基;
r9选自以下任意一种:
p是0、1、2、3或4;
r10选自:h、苄基或甲基;
r11选自:h或c1-c3烷基;
r12选自:h、c1-c3烷基或苄基;
r13选自:h、c1-c3烷基或苄基;
r14选自:h、c1-c6烷基或c1-c6烷氧羰基;
r15选自:h或c1-c4烷基;
或者,r8和r9与它们所连接的n原子一起形成一个环,选自以下任意一种:
s是0、1或2;
t是1、2或3;
q选自:s、o、nh、nch3、n(ch2)2oh、或chr17a;r17a选自:h、oh、羟基取代的c1-c3烷基或c(o)oh;
r16选自:h、c(o)oh、羟基取代的c1-c4烷基或c(o)nhso2r19;
r17选自:h、c(o)oh、羟基取代的c1-c4烷基、oh、c(o)或c(o)nhso2r19;
r18选自:c1-c4烷氧羰基、c1-c6烷基、c(o)oh、f、cl、br、i、oh、羟基取代的c1-c4烷基、nrarb或苯氧基羰基;其中,苯氧基羰基的苯基任选被f、cl、br、i、oh、cn、no2、nh2、cf3、cf2cf3、ocf3、ocf2cf3、so2nh2、c(o)oh、c(o)nh2或nhc(o)nh2取代;ra和rb独立地选自:h、c1-c4烷氧羰基或c1-c4烷基羰基;
r19选自:cf3、环丙基、c1-c4烷基、二甲基氨基或甲基取代的咪唑基;
r3、r4、r5、r6和r7各自独立地选自:h、rc、orc、src、s(o)rc、s(o)2rc、c(o)rc、c(o)oh、c(o)orc、oc(o)rc、nhrc、n(rc)2、c(o)nh2、c(o)nhrc、c(o)n(rc)2、nh(co)rc、nrc(co)rc、nh(co)orc、nrc(co)orc、nh(co)nh2、nh(co)nhrc、nh(co)n(rc)2、nrc(co)nhrc、nrc(co)n(rc)2、so2nh2、so2nhrc、so2n(rc)2、nhso2rc、nrcso2rc、nhso2nhrc、nhso2n(rc)2、nrcso2nhrc、nrcso2n(rc)2、c(o)nhnoh、c(o)nhnorc、c(o)nhso2rc、c(nh)nh2、c(nh)nhrc、c(nh)n(rc)2、f、cl、br、i、cn、no2、nh2、oh、cf3、cf2cf3、ocf3或ocf2cf3;
rc选自:苯基、杂芳基、环烷基、环烯基、杂环烷基、杂环烯基、取代或非取代的c1-c4烷基、烯基或炔基;
或者,r4、r5、r6和r7之中每两个与它们所连接到的原子一起形成取代或非取代的苯环、取代或非取代的杂芳环、取代或非取代的环烷烃环、取代或非取代的杂环烷烃环、或取代或非取代的杂环烯烃环;
w选自:h、f、cl、br、i、cn、no2、nh2、oh、cf3、cf2cf3、ocf3、ocf2cf3、c1-c4烷基、烯基、炔基、烷氧基、烷硫基、环烷基、卤代烷基、卤代烷氧基、卤代烷硫基、卤代环烷基或杂环烷基。
在某些实施方案中,所述苯并噁二唑类化合物包括其药学上可接受的盐、互变异构体、内消旋体、外消旋体、立体异构体、代谢产物、代谢前体、前药或溶剂化物。
在某些优选的实施方案中,所述苯并噁二唑类化合物中的r1为-(ch2)nar;其中,n是1;ar是取代或非取代的芳基或杂芳基;
r2为-(ch2)mnr8r9;其中,m是1;
r8选自:h或c1-c4烷基;
r9选自以下任意一种:
p是1或2;
r10是h;
r11选自:h或c1-c3烷基;
r12选自:h或c1-c3烷基;
r13选自:h或c1-c3烷基;
r14选自:h、c1-c6烷基或c1-c6烷氧羰基;
r15选自:h或c1-c4烷基;
或者,r8和r9与它们所连接的n原子一起形成一个环,选自以下任意一种:
s是0或1;
t是2或3;
q选自:s、o、nh、nch3、n(ch2)2oh或chr17a;r17a选自:h、oh、羟基取代的c1-c3烷基或c(o)oh;
r16选自:h、c(o)oh、羟基取代的c1-c4烷基或c(o)nhso2r19;
r17选自:h、c(o)oh、羟基取代的c1-c4烷基、oh、c(o)或c(o)nhso2r19;
r18选自:c1-c4烷氧羰基、c1-c6烷基、c(o)oh、f、cl、br、i、oh、羟基取代的c1-c4烷基、-nrarb或苯氧基羰基;苯氧基羰基的苯基任选被f、cl、br、i、oh、cn、no2、nh2、cf3、cf2cf3、ocf3、ocf2cf3、so2nh2、c(o)oh、c(o)nh2或nhc(o)nh2取代;ra和rb各自独立地选自:h、c1-c4烷氧羰基或c1-c4烷基羰基;
r19选自:cf3、环丙基、c1-c4烷基、二甲基氨基或甲基取代的咪唑基;
r3、r4、r5、r6和r7各自独立地选自:h、rc、orc、src、s(o)rc、s(o)2rc、c(o)rc、c(o)oh、c(o)orc、oc(o)rc、nhrc、n(rc)2、c(o)nh2、c(o)nhrc、c(o)n(rc)2、nh(co)rc、nrc(co)rc、nh(co)orc、nrc(co)orc、nh(co)nh2、nh(co)nhrc、nh(co)n(rc)2、nrc(co)nhrc、nrc(co)n(rc)2、so2nh2、so2nhrc、so2n(rc)2、nhso2rc、nrcso2rc、nhso2nhrc、nhso2n(rc)2、nrcso2nhrc、nrcso2n(rc)2、c(o)nhnoh、c(o)nhnorc、c(o)nhso2rc、c(nh)nh2、c(nh)nhrc、c(nh)n(rc)2、f、cl、br、i、cn、no2、nh2、oh、cf3、cf2cf3、ocf3或ocf2cf3;
rc选自:苯基、杂芳基、环烷基、环烯基、杂环烷基、杂环烯基、取代或非取代的c1-c4烷基、烯基或炔基;
或者,r4、r5、r6和r7之中每两个与它们所连接到的原子一起形成取代或非取代的苯环、取代或非取代的杂芳环、取代或非取代的环烷烃环、取代或非取代的杂环烷烃环、或取代或非取代的杂环烯烃环;
w选自:h、f、cl、br、i、cn、no2、nh2、oh、cf3、cf2cf3、ocf3、ocf2cf3、c1-c4烷基、烯基、炔基、烷氧基、烷硫基、环烷基、卤代烷基、卤代烷氧基、卤代烷硫基、卤代环烷基或杂环烷基。
进一步地,所述苯并噁二唑类化合物中的ar是取代或非取代的芳基或杂芳基,其选自:苯基、吡啶基、嘧啶基、喹啉基、异喹啉基、吲唑基、异噁唑基、噁二唑基、萘基、噻唑基或咪唑基;所述取代或非取代的芳基或杂芳基是未取代的或被一个或两个或三个独立地选自下列的取代基所取代:c1-c4烷基、c1-c4烷氧基、c1-c4烷氧羰基、c1-c4烷基磺酰基、羟基取代的c1-c4烷基、f、cl、br、i、cn、no2、nh2、oh、cf3、cf2cf3、ocf3、ocf2cf3、c(o)oh、c(o)nh2、吗啉基、哌啶基、四氢吡咯基、哌嗪基、n,n-二甲基氨基、四氢吡喃基、未取代或取代的苯基。
进一步地,所述苯并噁二唑类化合物中的rc选自:苯基、杂芳基、环烷基、环烯基、杂环烷基、杂环烯基、取代或非取代的c1-c4烷基、烯基或炔基;所述取代或非取代的c1-c4烷基是未取代的或被一个或两个或三个独立地各自选自下列的取代基所取代:rg、oh、(o)、c(o)oh、cn、nh2、f、cl、br、i、cf3、cf2cf3、nc(rh)(ri)、rj、orj、srj、s(o)rj、s(o)2rj、nhrj、n(rj)2、c(o)rj、c(o)nh2、c(o)nhrj、c(o)n(rj)2、nhc(o)rj、nrjc(o)rj、nhso2rj、nhc(o)orj、so2nhrj、so2n(rj)2、nhc(o)nh2、nhc(o)nhrj、吡咯烷-1-基、哌啶-1-基、哌嗪-1-基、4-甲基-哌嗪-1-基、吗啉-4-基、硫代吗啉-1,1-二氧代-4-基、nhc(o)ch(ch3)nhc(o)ch(ch3)nh或nhc(o)ch(ch3)nhc(o)-ch(ch3)nhrj;
rg选自2~5个碳的螺烷基,其每个是未取代的或被oh、(o)、cn、nh2、f、cl、br、i、cf3、cf2cf3、nh(ch3)或n(ch3)2取代;
rh和ri选自独立选择的烷基,或者与它们所连接到的n一起是氮丙啶-1-基、氮杂环丁烷-1-基、吡咯烷-1-基或哌啶-1-基,每个具有一个未被替代的或被o、c(o)、cnoh、cnoch3、s、s(o)、s(o)2或nh替代的ch2部分;
rj选自:苯基、杂芳基、环烷基、环烯基、杂环烷基、杂环烯基、c1-c4烷基、烯基或炔基。
作为更优选的实施方案,本发明所述的苯并噁二唑类化合物或其药学上可接受的盐、互变异构体、内消旋体、外消旋体、立体异构体、代谢产物、代谢前体、前药或溶剂化物为如下表1所示的化合物:
表1化合物的结构与命名
本发明的苯并噁二唑类化合物可作为药用盐使用。该盐可为下列酸中的至少一种的酸盐:半乳糖二酸、d-葡糖醛酸、甘油磷酸、马尿酸、羟乙磺酸、乳糖酸、马来酸、1,5-萘二磺酸、萘-2-磺酸、新戊酸、对苯二甲酸、硫氰酸、胆酸、正十二烷基硫酸、苯磺酸、柠檬酸、d-葡萄糖,乙醇酸、乳酸、苹果酸、丙二酸、扁桃酸、磷酸、丙酸、盐酸、硫酸、酒石酸、琥珀酸、甲酸、氢碘酸、氢溴酸、甲烷磺酸、烟酸、硝酸、乳清酸、草酸、苦味酸、l-焦谷氨酸、糖精酸、水杨酸、龙胆酸、对甲苯磺酸、戊酸、棕榈酸、葵二酸、硬脂酸、月桂酸、乙酸、己二酸、碳酸、4-苯磺酸、乙烷二磺酸、乙基琥珀酸、富马酸、3-羟基萘-2-甲酸、1-羟基萘-2-甲酸、油酸、十一碳烯酸、抗坏血酸、樟脑酸、樟脑磺酸、二氯乙酸、乙烷磺酸。另一方面,该盐也可以是本发明的化合物与金属(包括钠、钾、钙等)离子或药学上可接受的胺(包括乙二胺、氨丁三醇等)或铵离子形成的盐。
本发明还提供了所述苯并噁二唑类化合物或其药学上可接受的盐、互变异构体、内消旋体、外消旋体、立体异构体、代谢产物、代谢前体、前药或溶剂化物在制备具有pd-1/pd-l1抑制活性的抑制剂中的应用。
本发明人发现,式(i)所示的苯并噁二唑类化合物是免疫检查点pd-1/pd-l1的抑制剂。均相时间分辨荧光实验等研究结果表明,式(i)化合物对pd-1和pd-l1的相互作用具有很好的抑制作用,且能显著地增加t细胞的增殖能力,促进免疫因子的生成,并激活t细胞的抗肿瘤免疫活性,因而可用于肿瘤的免疫治疗。因此,本发明的苯并噁二唑类化合物,包括其药学上可接受的盐、互变异构体、内消旋体、外消旋体、立体异构体、代谢产物、代谢前体、前药或溶剂化物作为免疫检查点抑制剂可用于制备肿瘤免疫治疗药物。
本发明的苯并噁二唑类化合物可用于肿瘤的免疫治疗。所述肿瘤包括但不限于:骨癌、急性骨髓性白血病、慢性骨髓性白血病、急性淋巴细胞系白血病、慢性淋巴细胞系白血病、骨髓增生性疾病、多发性骨髓瘤、骨髓增生异常综合征、霍奇金氏淋巴瘤、非霍奇金氏淋巴瘤、血管瘤、肉芽瘤、黄瘤、脑膜肉瘤、神经胶质瘤、星形细胞瘤、成神经管细胞瘤、室管膜瘤、生殖细胞瘤(松果体瘤)、多形性成胶质细胞瘤、少突神经胶质细胞瘤、神经鞘瘤、成视网膜细胞瘤、纤维神经瘤、肉瘤、食道癌、胃癌、胰腺癌、大肠癌、结肠癌、直肠癌、肾癌、前列腺癌、淋巴癌、睾丸癌、间质细胞癌、肺癌、肝癌、皮肤癌、恶性黑素瘤和基底细胞癌等。
在某些实施方案中,本发明的式(i)化合物可单独使用。而在某些实施方案中,本发明的式(i)化合物可与一种或多种其他抗肿瘤药物联合使用。可选择与本发明的苯并噁二唑类化合物联合使用的抗肿瘤药物包括但不限于:化疗药物,如多西他赛、紫杉醇、abraxane、多柔比星、奥沙利铂、卡铂、顺铂、伊立替康、吉西他滨和环磷酰胺等;免疫检查点抑制剂,如ctla-4抑制剂(如yervoy)、pd-1抑制剂(如keytruda和opdivo)、pd-l1抑制剂(如tecentriq)、lag-3抑制剂(如bms986016、regn3767和lag525)、tim-3抑制剂(如tsr-022和mbg-453)、tigit抑制剂(如mtig7192a)、b7h4抑制剂和vista抑制剂(如jnj-61610588)等;抗体药物缀合物(adc),如kadcyla等;激酶抑制剂,如shp-2抑制剂、b-raf抑制剂(如vemurafenib)、mek抑制剂(如cobimetinib)和btk抑制剂(如ibrutinib)等;ido抑制剂(如epacadostat)等。
本发明还提供了一种用于肿瘤免疫治疗的药物组合物,其中含有如本发明所述的治疗有效量的式(i)所示苯并噁二唑类化合物或其药学上可接受的盐、互变异构体、内消旋体、外消旋体、立体异构体、代谢产物、代谢前体、前药或溶剂化物作为活性成分和药学上可接受的载体。可任意混合的载体根据剂型、给药形式等可以改变。载体的例子包括赋形剂、粘合剂、崩解剂、润滑剂、矫味剂、香味剂、着色剂和甜味剂等。所述药物组合物可以是胶囊剂、散剂、片剂、颗粒剂、丸剂、注射剂、糖浆剂、口服液、吸入剂、软膏剂、栓剂和贴剂等制剂学上常规的制剂形式。
进一步地,所述药物组合物是胶囊剂、散剂、片剂、颗粒剂、丸剂、注射剂、糖浆剂、口服液、吸入剂、软膏剂、栓剂或贴剂。
为了使片剂形式的药物组合物成形,可使用本领域任何已知并广泛使用的赋形剂。例如,载体,如乳糖、白糖、氯化钠、葡萄糖、尿素、淀粉、碳酸钙、高岭土、结晶纤维素和硅酸等;粘合剂,如水、乙醇、丙醇、普通糖浆、葡萄糖溶液、淀粉溶液、明胶溶液,羧甲基纤维素、紫胶、甲基纤维素和磷酸钾、聚乙烯吡咯烷酮等;崩解剂,如干淀粉、藻酸钠、琼脂粉和海带粉,碳酸氢钠、碳酸钙、聚乙烯脱水山梨醇的脂肪酸酯、十二烷基硫酸钠、单硬脂酸甘油酯、淀粉和乳糖等;崩解抑制剂,如白糖、甘油三硬脂酸酯、椰子油和氢化油;吸附促进剂,如季胺碱和十二烷基硫酸钠等;润湿剂,如甘油、淀粉等;吸附剂,如淀粉、乳糖、高岭土、膨润土和胶体硅酸等;以及润滑剂,如纯净的滑石,硬脂酸盐、硼酸粉和聚乙二醇等。还可以根据需要选用通常的涂渍材料制成糖衣片剂、涂明胶膜片剂、肠衣片剂、涂膜片剂、双层膜片剂及多层片剂。
为了使丸剂形式的药物组合物成形,可使用本领域任何已知的并广泛使用的赋形剂,例如,载体,如乳糖,淀粉,椰子油,硬化植物油,高岭土和滑石粉等;粘合剂,如阿拉伯树胶粉,黄蓍胶粉,明胶和乙醇等;崩解剂,如琼脂和海带粉等。
为了使栓剂形式的药物组合物成形,可使用本领域任何已知并广泛使用的赋性剂,例如,聚乙二醇,椰子油,高级醇,高级醇的酯,明胶和半合成的甘油酯等。
为了制备针剂形式的药物组合物,可将溶液或悬浮液消毒后,制成与血液等渗压的针剂。在制备针剂时,也可使用本领域内任何常用的载体。例如,水,乙醇,丙二醇,乙氧基化的异硬脂醇,聚氧基化的异硬脂醇和聚乙烯脱水山梨醇的脂肪酸酯等。此外,还可加入通常的溶解剂、缓冲剂和止痛剂等。
所述药物组合物中,所述稀释剂可为本领域中常规的稀释剂。
所述的药物组合物可以是口服的形式,也可以是无菌注射水溶液形式,可按照本领域任何已知制备药用组合物的方法制备口服或注射组合物。
本发明的苯并噁二唑类化合物的制备可参照实施例中的方法进行,特别地,当所述式(i)化合物中的r2为-(ch2)mnr8r9,且m是1时,本发明的苯并噁二唑类化合物的制备可参照以下合成路线或改进的方法进行。
在上述合成路线中,y是溴、氯、碘、对甲苯磺酸酯、甲磺酸酯或三氟甲磺酸酯;r1、r3、r4、r5、r6、r7、r8、r9和w的定义与所述式(i)化合物中的定义一致。所述化合物的合成具体包括以下步骤:
(1)由化合物m-1与化合物m-2在碱的作用下反应得到化合物m-3:所采用的溶剂包括但不限于:苯、甲苯、氯仿、正己烷、环己烷、二氯甲烷、1,2-二氯乙烷、甲基叔丁基醚、乙酸乙酯、乙酸丙酯、乙酸丁酯、甲醇、乙醇、丙酮、四氢呋喃、乙醚、乙腈、n,n-二甲基甲酰胺、二甲亚砜或者用这些溶剂任选组成的混合溶剂,优选丙酮、n,n-二甲基甲酰胺、四氢呋喃、乙腈或二氯甲烷;所采用的碱包括但不限于:三乙胺、二异丙基乙胺、1,8-二氮杂环[5,4,0]十一烯-7、碳酸钾、碳酸钠、碳酸氢钾或碳酸氢钠;反应温度为0℃至150℃,优选温度为50℃至100℃;
(2)由化合物m-3与叠氮化物反应得到化合物m-4:所采用的溶剂包括但不限于:苯、甲苯、氯仿、正己烷、环己烷、二氯甲烷、1,2-二氯乙烷、甲基叔丁基醚、乙酸乙酯、乙酸丙酯、乙酸丁酯、甲醇、乙醇、丙酮、四氢呋喃、乙醚、乙腈、n,n-二甲基甲酰胺、二甲亚砜或者用这些溶剂任选组成的混合溶剂,优选丙酮、n,n-二甲基甲酰胺、四氢呋喃或乙腈;所采用的叠氮化物包括但不限于:叠氮化钠或叠氮化钾;反应温度为0℃至150℃,优选温度为50℃至100℃;
(3)由化合物m-4经环合反应得到化合物m-5:所采用的溶剂包括但不限于:苯、甲苯、氯仿、正己烷、环己烷、二氯甲烷、1,2-二氯乙烷、甲基叔丁基醚、乙酸乙酯、乙酸丙酯、乙酸丁酯、甲醇、乙醇、丙酮、四氢呋喃、乙腈、n,n-二甲基甲酰胺、二甲亚砜或者用这些溶剂任选组成的混合溶剂,优选丙酮、n,n-二甲基甲酰胺、四氢呋喃、甲苯或乙腈;反应温度为室温至200℃,优选温度为50℃至150℃;
(4)由化合物m-5与r1oh在碱的作用下反应得到化合物m-6:所采用的溶剂包括但不限于:苯、甲苯、氯仿、正己烷、环己烷、二氯甲烷、1,2-二氯乙烷、甲基叔丁基醚、乙酸乙酯、乙酸丙酯、乙酸丁酯、丙酮、四氢呋喃、乙腈、n,n-二甲基甲酰胺、二甲亚砜或者用这些溶剂任选组成的混合溶剂,优选丙酮、n,n-二甲基甲酰胺、四氢呋喃或乙腈;所采用的碱包括但不限于:三乙胺、二异丙基乙胺、1,8-二氮杂环[5,4,0]十一烯-7、碳酸钾、碳酸钠、碳酸氢钾或碳酸氢钠;反应温度为-20℃至100℃,优选温度为室温至50℃;
(5)由化合物m-6在脱氧剂的作用下反应得到化合物m-7:所采用的溶剂包括但不限于:苯、甲苯、氯仿、正己烷、环己烷、二氯甲烷、1,2-二氯乙烷、甲基叔丁基醚、乙酸乙酯、乙酸丙酯、乙酸丁酯、丙酮、甲醇、乙醇、四氢呋喃、乙腈、n,n-二甲基甲酰胺、二甲亚砜或者用这些溶剂任选组成的混合溶剂,优选n,n-二甲基甲酰胺、四氢呋喃或乙腈;所采用的脱氧剂包括但不限于:三苯基膦、三乙基膦、三丁基膦或三环己基膦;反应温度为0℃至150℃,优选温度为50℃至120℃;
(6)由化合物m-7与nhr8r9进行还原胺化反应得到化合物(ia):所采用的溶剂包括但不限于:乙酸、苯、甲苯、氯仿、正己烷、环己烷、二氯甲烷、1,2-二氯乙烷、甲基叔丁基醚、乙酸乙酯、乙酸丙酯、乙酸丁酯、甲醇、乙醇、四氢呋喃、乙腈、n,n-二甲基甲酰胺、二甲亚砜或者用这些溶剂任选组成的混合溶剂;所采用的还原剂包括但不限于:硼氢化钠、三乙酰氧基硼氢化钠或氰基硼氢化钠;反应温度为-20℃至80℃,优选温度为0℃至50℃;
(7)或者,由化合物m-7经还原反应得到化合物m-8,所采用的还原剂包括但不限于:硼氢化钠或硼氢化钾;m-8再经卤化或磺酸酯化得到化合物m-9,所采用的卤化剂包括但不限于:pph3/cbr4、pbr3、pobr3、hbr、pcl3、pocl3、hcl或hi,所采用的磺酸酯化试剂(在碱的作用下)包括但不限于:对甲苯磺酰氯、甲磺酰氯、甲磺酸酐、三氟甲磺酰氯或三氟甲磺酸酐;m-9在碱的作用下与nhr8r9进行烷基化反应得到化合物(ia),所采用的碱包括但不限于:三乙胺、二异丙基乙胺、1,8-二氮杂环[5,4,0]十一烯-7、碳酸钾、碳酸钠、碳酸氢钾或碳酸氢钠。
本发明的式(i)化合物或其药学上可接受的盐、互变异构体、内消旋体、外消旋体、立体异构体、代谢产物、代谢前体、前药或溶剂化物的用量可根据患者年龄、体重、症状和给药途径等而适当改变。对成人而言,在口服给药时,一次给药量的下限是0.1mg(优选1mg),上限是1000mg(优选500mg);在静脉给药时,一次给药量的下限是0.01mg(优选0.1mg),上限是500mg(优选250mg)。也可根据疾病程度的不同和剂型的不同而偏离此剂量范围。
有益效果:与现有技术相比,本发明具有如下优点:
(1)本发明的新型苯并噁二唑类化合物可以显著抑制pd-1/pd-l1的相互作用,且活性显著优于已知的pd-1/pd-l1抑制剂bms-1016。尤其重要的是,本发明的苯并噁二唑类化合物可以显著地阻断pd-l1对t细胞的抑制作用,并可阻断肿瘤细胞抑制t细胞增殖和分泌ifn-γ的效应,因而具有增强t细胞抗肿瘤免疫的功效。因此,本发明的化合物作为免疫检查点pd-1/pd-l1抑制剂可用于制备肿瘤免疫治疗的药物。
(2)本发明的苯并噁二唑类化合物的合成路线设计巧妙、简便易行,原料便宜易得,合成工艺安全、环保,易于规模化生产。
(3)本发明的苯并噁二唑类化合物作为新型pd-1/pd-l1小分子抑制剂,比现有的pd-1/pd-l1单抗类药物具有成本低廉、易于制备及潜在的免疫反应副作用小等优点,其即可单独使用,也可与化疗药或其他肿瘤免疫治疗药物联用,有望成为新型肿瘤免疫治疗药物。
附图说明
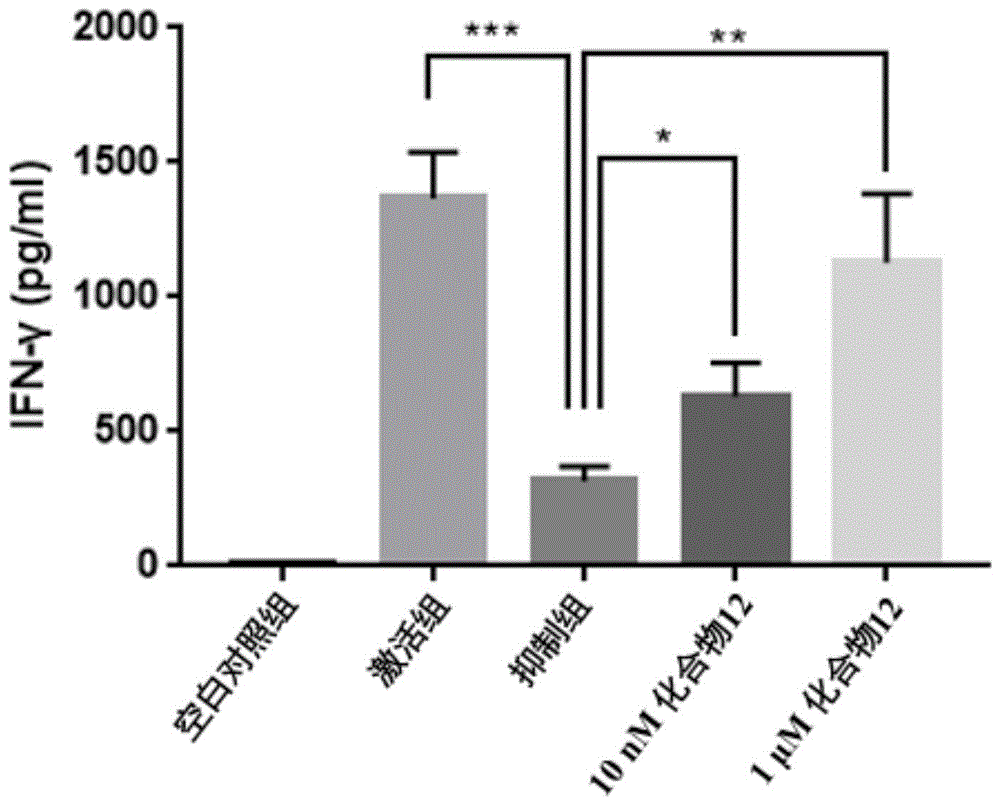
图1为苯并噁二唑类化合物阻断pd-l1抑制人外周血单个核细胞(pbmc)分泌ifn-γ的效应图:空白对照组、激活组、抑制组以及不同浓度的化合物组的人pbmc在48小时后ifn-γ的分泌情况(n=3,*p<0.05,**p<0.01,***p<0.001);
图2为苯并噁二唑类化合物阻断肿瘤细胞抑制人t细胞增殖的效应图:空白对照组、激活组、抑制组以及不同浓度的化合物组的t细胞在48小时后的增殖情况;
图3为苯并噁二唑类化合物阻断肿瘤细胞抑制人t细胞分泌ifn-γ的效应图:空白对照组、激活组、抑制组以及不同浓度的化合物组的t细胞在48小时后ifn-γ的分泌情况(n=3,*p<0.05,**p<0.01,***p<0.001)。
具体实施方式
下面通过实施例具体说明本发明的内容。在本发明中,以下所述的实施例是为了更好的阐述本发明,并不是用来限制本发明的范围。在不背离本发明的精神和范围的前提下,可以对本发明进行各种变化和修饰。
实施例1
n-((5-((3-氰基苄基)氧基)-7-((2-甲基-[1,1'-联苯]-3-基)甲氧基)苯并[c][1,2,5]噁二唑-4-基)甲基)-d-丝氨酸(化合物1)
合成路线:
化合物i-2的合成
取化合物m-2(1.8g)溶于无水丙酮(30ml)中,加入无水碳酸氢钠(1.3g)和四丁基碘化胺(tbai,300mg),并缓慢加入i-1(2g),氩气保护,70℃油浴加热。反应约6小时后停止加热,减压蒸除溶剂,加入乙酸乙酯(25ml)稀释,用水(10mlx3)和饱和食盐水(10mlx2)洗涤,无水硫酸钠干燥。减压蒸除溶剂,残余物经柱层析(洗脱剂:石油醚/乙酸乙酯=3:1)纯化,得淡黄色固体化合物i-2(2.14g)。
化合物i-3的合成
取化合物i-2(2.33g)溶于乙腈(50ml)中,分批缓慢加入叠氮化钠(493mg),75℃油浴加热。反应约5小时后停止加热,减压蒸除溶剂,得黄色固体化合物i-3的粗品(2.78g)。
化合物i-4的合成
取化合物i-3的粗品(2.78g)溶于甲苯(30ml),置于110℃油浴加热。反应约5小时后停止加热,静置2小时,有固体析出,抽滤,得黄色固体化合物i-4(1.2g)。
化合物i-5的合成
取化合物i-4(500mg)加入乙腈(15ml)中,加入无水碳酸钾(364mg)和3-羟甲基苯甲腈(176mg),反应液在常温反应12小时后停止反应,减压蒸除溶剂,加入乙酸乙酯(25ml)稀释,用水(10mlx3)和饱和食盐水(10mlx2)洗涤,无水硫酸钠干燥。减压蒸除溶剂,残余物经柱层析(洗脱剂:石油醚/乙酸乙酯=1:1)纯化,得黄色固体化合物i-5(449mg)。
化合物i-6的合成
取化合物i-5(449mg)溶于n,n-二甲基甲酰胺(5ml),加入三苯基膦(240mg),90℃油浴加热。反应约6小时后停止加热,加入水(10ml),用乙酸乙酯(10mlx3)萃取,无水硫酸钠干燥。减压蒸除溶剂,残余物经柱层析(洗脱剂:石油醚/乙酸乙酯=2:1)纯化,得黄色固体化合物i-6(159mg)。
化合物i-7的合成
取d-丝氨酸乙酯盐酸盐(214mg)溶于二氯甲烷与甲醇的混合溶剂(3:1,4ml),加入n,n-二异丙基乙胺(163mg)室温搅拌20分钟,加入化合物i-6(150mg)和三乙酰氧基硼氢化钠(stab,267mg),室温反应12小时后停止反应。减压蒸除溶剂,残余物经柱层析(洗脱剂:石油醚/乙酸乙酯=1:1)纯化,得黄色油状化合物i-7(135mg)。
化合物1的合成
取化合物i-7(135mg)溶于甲醇与四氢呋喃的混合溶剂(2:1,3ml),加入一水合氢氧化锂(24mg),室温反应5小时后,减压蒸除溶剂,残余物经柱层析(洗脱剂:甲醇/水=1:1)纯化,得淡黄色固体化合物1(46mg):1hnmr(300mhz,dmso)δ8.04(s,1h),7.95(d,j=7.9hz,1h),7.84(d,j=7.7hz,1h),7.64(t,j=7.8hz,1h),7.54(d,j=7.4hz,1h),7.51–7.42(m,2h),7.39(d,j=6.8hz,1h),7.37–7.29(m,3h),7.29–7.17(m,2h),5.46(s,2h),5.44(s,2h),3.89(s,2h),3.79(s,1h),3.45(dd,j=9.6,5.4hz,1h),3.19(t,j=9.5hz,1h),2.60(dd,j=9.2,5.3hz,1h),2.24(s,3h).esi-ms:m/z587.18957[m+na]+。
实施例2
n-(2-(((5-((3-氰基苄基)氧基)-7-((2-甲基-[1,1'-联苯]-3-基)甲氧基)苯并[c][1,2,5]噁二唑-4-基)甲基)氨基)乙基)乙酰胺(化合物2)
参照实施例1的方法,将实施例1中的d-丝氨酸乙酯盐酸盐替换成n-乙酰基乙二胺,制得化合物2:1hnmr(300mhz,cdcl3)δ7.73(s,1h),7.71–7.60(m,2h),7.54(t,j=7.7hz,1h),7.40(m,j=13.3,6.7hz,4h),7.29(m,4h),6.58(s,1h),6.28(s,1h),5.38(s,2h),5.21(s,2h),4.09(s,2h),3.34(dd,j=10.8,5.3hz,2h),2.74(t,j=5.6hz,2h),2.28(s,3h),1.95(s,3h).hrms(esi):m/z562.24496[m+h]+。
实施例3
n-((5-((3-氰基苄基)氧基)-7-((2-甲基-[1,1'-联苯]-3-基)甲氧基)苯并[c][1,2,5]噁二唑-4-基)甲基)甘氨酸(化合物3)
参照实施例1的方法,将实施例1中的d-丝氨酸乙酯盐酸盐替换成甘氨酸甲酯盐酸盐,制得化合物3:1hnmr(300mhz,cd3od)δ7.95–7.83(m,2h),7.75–7.54(m,3h),7.50–7.21(m,7h),6.96(s,1h),5.44(s,2h),5.40(s,2h),4.05(s,2h),3.18(s,2h),2.27(s,3h).ms(esi):m/z533.3[m-h]+。
实施例4
n-((5-((4-氰基苄基)氧基)-7-((2-甲基-[1,1'-联苯]-3-基)甲氧基)苯并[c][1,2,5]噁二唑-4-基)甲基)-d-丝氨酸(化合物4)
参照实施例1的方法,将实施例1中的3-羟甲基苯甲腈替换成4-羟甲基苯甲腈,制得化合物4:1hnmr(500mhz,cd3od)δ7.77(d,j=7.8hz,2h),7.71(d,j=7.8hz,2h),7.51–7.43(m,3h),7.39(t,j=7.1hz,1h),7.33(d,j=7.4hz,2h),7.31–7.27(m,2h),7.00(s,1h),5.49(s,2h),5.47(s,2h),4.35–4.19(m,2h),3.86–3.74(m,2h),3.28(t,1h).hrms(esi):m/z587.18979[m+h]+。
实施例5
n-((5-(吡啶-3-基甲氧基)-7-((2-甲基-[1,1'-联苯]-3-基)甲氧基)苯并[c][1,2,5]噁二唑-4-基)甲基)-d-丝氨酸(化合物5)
参照实施例1的方法,将实施例1中的3-羟甲基苯甲腈替换成3-吡啶甲醇,制得化合物5:1hnmr(500mhz,cd3od)δ8.75(s,1h),8.56(s,1h),8.07(d,j=6.6hz,1h),7.52(d,j=6.1hz,2h),7.46(t,j=7.2hz,2h),7.39(t,j=7.1hz,1h),7.33–7.27(m,4h),7.07(s,1h),5.50(s,2h),5.45(s,2h),4.20(s,2h),3.79(s,2h),3.27–3.19(m,1h).hrms(esi):m/z563.18947[m+na]+。
实施例6
n-((5-(3-氟苄基)氧基)-7-((2-甲基-[1,1'-联苯]-3-基)甲氧基)苯并[c][1,2,5]噁二唑-4-基)甲基)-d-丝氨酸(化合物6)
参照实施例1的方法,将实施例1中的3-羟甲基苯甲腈替换成3-氟苄醇,制得化合物6:1hnmr(300mhz,dmso)δ7.67–6.96(m,13h),5.44(s,2h),5.39(s,2h),3.99(d,j=16.8hz,1h),3.92(d,j=12.5hz,1h),3.48(s,1h),2.79(s,1h),2.22(s,3h),1.99(s,1h).hrms(esi):m/z580.18710[m+na]+。
实施例7
n-((5-((3-氯苄基)氧基)-7-((2-甲基-[1,1'-联苯]-3-基)甲氧基)苯并[c][1,2,5]噁二唑-4-基)甲基)-d-丝氨酸(化合物7)
参照实施例1的方法,将实施例1中的3-羟甲基苯甲腈替换成3-氯苄醇,制得化合物7:1hnmr(300mhz,cd3od)δ7.70–7.57(m,2h),7.57–7.53(m,1h),7.48–7.34(m,6h),7.34–7.22(m,4h),6.93(s,1h),5.40(s,2h),5.34(s,2h),4.13(d,j=13.5hz,1h),4.07(d,j=13.4hz,1h),3.73(dd,j=10.7,5.6hz,1h),3.66(dd,j=10.6,6.0hz,1h),3.16(t,j=5.8hz,1h),2.26(s,3h).ms(esi):m/z596.3[m+na]+。
实施例8
n-((5-((5-氰基吡啶-3-基)甲氧基)-7-((2-甲基-[1,1'-联苯]-3-基)甲氧基)苯并[c][1,2,5]噁二唑-4-基)甲基)-d-丝氨酸(化合物8)
参照实施例1的方法,将实施例1中的3-羟甲基苯甲腈替换成5-(羟甲基)烟腈,制得化合物8:1hnmr(300mhz,dmso)δ9.08(s,1h),9.01(s,1h),8.48(s,1h),7.64–7.19(m,9h),5.54(s,2h),5.49(s,2h),4.13–3.96(m,2h),3.53–3.46(m,2h),3.05(t,1h),2.25(s,3h).ms(esi):m/z588.3[m+na]+。
实施例9
n-((5-((2-氰基吡啶-4-基)甲氧基)-7-((2-甲基-[1,1'-联苯]-3-基)甲氧基)苯并[c][1,2,5]噁二唑-4-基)甲基)-d-丝氨酸(化合物9)
参照实施例1的方法,将实施例1中的3-羟甲基苯甲腈替换成2-氰基-4-羟甲基吡啶,制得化合物9:1hnmr(300mhz,meod)δ8.72(d,j=4.9hz,1h),8.27(s,1h),7.91–7.80(m,1h),7.48–7.38(m,3h),7.35(t,j=7.2hz,1h),7.31–7.25(m,2h),7.25–7.18(m,2h),6.98(s,1h),5.60(s,2h),5.47(s,2h),4.60(s,1h),4.54(s,1h),4.02(s,1h),3.86(s,1h),3.61(s,1h),2.26(d,j=8.2hz,3h).hrms(esi):m/z566.18910[m+h]+。
实施例10
n-((5-((3-氰基苄基)氧基)-7-((3-(2,3-二氢苯并[b][1,4]二氧杂环己烯-6-基)-2-甲基苄基)氧基)苯并[c][1,2,5]噁二唑-4-基)甲基)-d-丝氨酸(化合物10)
合成路线:
化合物a-2的合成
取化合物a-1(5g)溶于甲醇(60ml)中,于室温下缓慢滴加浓硫酸(10ml),约15分钟加毕,85℃油浴加热。反应约10小时后停止加热,减压蒸除溶剂,用乙酸乙酯(50mlx3)萃取,饱和碳酸钠(30mlx3)和饱和食盐水(25mlx2)洗涤,无水硫酸钠干燥。减压蒸除溶剂,残余物经柱层析(洗脱剂:石油醚/乙酸乙酯=50:1)纯化,得淡黄色油状化合物a-2(5.1g)。
化合物a-3的合成
取化合物a-2(3g),苯并二氧六环-4-硼酸酯(3.8g),[1,1’-双(二苯基膦)二茂铁]二氯化钯二氯甲烷络合物(530mg),无水碳酸钾(3.5g),18-冠-6-醚(345mg),加入到100ml三颈瓶中,氩气保护,换气三次。室温下加入甲苯(21ml),无水乙醇(7ml),水(3.5ml)。90℃油浴加热,反应约5小时后停止加热,减压蒸除溶剂,用乙酸乙酯25ml稀释,硅藻土抽滤,母液用乙酸乙酯(45mlx3)萃取,饱和食盐水(30mlx2)洗涤,无水硫酸钠干燥。减压蒸除溶剂,残余物经柱层析(洗脱剂:石油醚/乙酸乙酯=8:1)纯化,得淡黄色油状化合物a-3(3.96g)。
化合物a-4的合成
取化合物a-3(3.96g)溶于甲醇(15ml)和四氢呋喃(15ml)的混合溶剂中,于冰浴下缓慢加入一水合氢氧化锂(1.5g),室温下反应约6小时后停止反应,减压蒸除溶剂。1n盐酸酸化调ph=2~3,析出白色固体化合物a-4(3.3g)。
化合物a-5的合成
取化合物a-4(3.96g),溶于无水四氢呋喃(45ml)中,冰盐浴下缓慢滴加硼烷四氢呋喃复合物(29ml),室温下反应约5小时停止反应,冰浴下加水淬灭反应。用乙酸乙酯(50mlx3)萃取,和饱和食盐水(25mlx2)洗涤,无水硫酸钠干燥。减压蒸除溶剂,得白色固体化合物a-5(3.0g)。
化合物a-6的合成
取化合物a-5(3.0g),三苯基膦(6.2g),溶于无水四氢呋喃(30ml)中,于冰浴下缓慢加入四溴化碳(7.8g),室温下反应约4小时后停止反应。减压蒸除溶剂,用二氯甲烷(30ml)稀释后硅藻土抽滤,母液经柱层析(洗脱剂:石油醚/乙酸乙酯=5:1)纯化,得淡黄色油状化合物a-6(3.6g)。
化合物ii-1的合成
取化合物m-2(521mg)溶于无水丙酮(10ml)中,加入无水碳酸氢钠(635mg)和四丁基碘化胺(tbai,100mg),并缓慢加入a-6(640mg),氩气保护,70℃油浴加热。反应约6小时后停止加热,减压蒸除溶剂,加入乙酸乙酯(10ml)稀释,用水(10mlx3)和饱和食盐水(10mlx2)洗涤,无水硫酸钠干燥。减压蒸除溶剂,残余物经柱层析(洗脱剂:石油醚/乙酸乙酯=3:1)纯化,得淡黄色固体化合物ii-1(854mg)。
化合物10的合成
参照实施例1的方法,将实施例1中的化合物i-2替换成化合物ii-1,制得化合物10:1hnmr(300mhz,cd3od)δ7.91(d,j=1.5hz,2h),7.77–7.52(m,4h),7.44(dd,j=6.7,1.9hz,1h),7.27–7.19(m,2h),6.95(s,1h),6.89(d,j=7.9hz,1h),6.78–6.71(m,2h),5.43(s,2h),5.40(s,2h),4.29(s,5h),4.15(d,j=13.5hz,1h),4.09(d,j=13.5hz,1h),3.74(dd,j=10.7,5.6hz,1h),3.65(dd,j=10.7,6.2hz,1h),3.36(d,j=4.9hz,1h),3.17(t,j=5.9hz,1h).hrms(esi):m/z623.21392[m+h]+。
实施例11
n-((5-((2-氰基吡啶-4-基)甲氧基)-7-((3-(2,3-二氢苯并[b][1,4]二氧杂环己烯-6-基)-2-甲基苄基)氧基)苯并[c][1,2,5]噁二唑-4-基)甲基)-d-丝氨酸(化合物11)
参照实施例10的方法,将实施例10中的3-羟甲基苯甲腈替换成2-氰基-4-羟甲基吡啶,制得化合物11:1hnmr(300mhz,cd3od)δ8.69(d,j=5.0hz,1h),8.02(s,1h),7.72(d,j=4.3hz,1h),7.38(dd,j=5.8,3.2hz,1h),7.20(dd,j=8.2,5.5hz,2h),6.94–6.83(m,2h),6.79–6.68(m,2h),5.46(s,2h),5.40(s,2h),4.28(s,4h),4.21(d,j=13.5hz,1h),4.14(d,j=13.5hz,1h),3.75(dd,j=10.7,5.5hz,1h),3.66(dd,j=10.7,6.1hz,1h),3.19(t,j=5.8hz,1h),2.25(s,3h).ms(esi):m/z624.2[m+na]+。
实施例12
n-((5-(吡啶-3-基甲氧基)-7-((2-溴-[1,1'-联苯]-3-基)甲氧基)苯并[c][1,2,5]噁二唑-4-基)甲基)-l-丝氨酸(化合物12)
合成路线:
化合物b-2的合成
取化合物b-1(4.2g,2.8ml)溶于水(10ml)中,冰盐浴下缓慢加入盐酸(6ml),搅拌5分钟,缓慢滴加亚硝酸钠(1.88g)的水溶液,20分钟加毕,冰盐浴下搅拌30分钟后,向反应液中缓慢滴加碘化钾(7.5g)的水溶液,室温下反应8小时后停止反应。加入硫代硫酸钠的饱和水溶液(35ml),用乙酸乙酯(50mlx3)萃取,饱和食盐水(25mlx3)洗涤,无水硫酸钠干燥。减压蒸除溶剂,残余物经柱层析(洗脱剂:石油醚)纯化,得淡黄色油状化合物b-2(5.2g)
化合物b-3的合成
取苯硼酸(1.08g),四(三苯基膦)钯(465mg),无水碳酸钠(1.28g)置于三颈瓶中,氩气保护。将化合物b-2(2.39g)溶于甲苯(8ml)中,加入反应瓶,加入甲苯(22ml),水(3ml),80℃油浴加热。反应约4小时后停止加热,减压蒸除溶剂,用乙酸乙酯(15ml)稀释,硅藻土抽滤,母液经柱层析(洗脱剂:石油醚)纯化,得淡黄色油状化合物b-3(1.78g)。
化合物b-4的合成
取化合物b-3(1.6g),溶于四氯化碳(50ml)中,加入n-溴代丁二酰亚胺(1.21g),分批加入过氧化苯甲酰(10mg),90℃油浴加热。反应约6小时后停止加热,减压蒸除溶剂,残余物经柱层析(洗脱剂:石油醚)纯化,得无色油状化合物b-4(1.43g)。
化合物iii-1的合成
取化合物m-2(638mg)溶于无水丙酮(10ml)中,加入无水碳酸氢钠(405mg)和四丁基碘化胺(tbai,200mg),并缓慢加入b-4(800mg),氩气保护,70℃油浴加热。反应约6小时后停止加热,减压蒸除溶剂,加入乙酸乙酯(15ml)稀释,用水(10mlx3)和饱和食盐水(10mlx2)洗涤,无水硫酸钠干燥。减压蒸除溶剂,残余物经柱层析(洗脱剂:石油醚/乙酸乙酯=5:1)纯化,得淡黄色固体化合物iii-1(726mg)。
化合物12的合成
参照实施例1的方法,将实施例1中的i-2替换成iii-1,将3-羟甲基苯甲腈替换成3-吡啶甲醇,并将d-丝氨酸乙酯盐酸盐替换成l-丝氨酸乙酯盐酸盐,制得化合物12:1hnmr(300mhz,cd3od)δ8.69(s,1h),8.53(d,j=4.0hz,1h),8.12(d,j=7.9hz,1h),7.66(d,j=6.4hz,1h),7.57–7.32(m,8h),6.97(s,1h),5.54(s,2h),5.42(s,2h),4.14(d,j=13.5hz,1h),4.08(d,j=13.5hz,1h),3.74(dd,j=10.7,5.6hz,1h),3.65(dd,j=10.6,6.3hz,1h),3.16(t,j=5.9hz,1h).hrms(esi):m/z627.08574[m+na]+。
实施例13
n-((5-(吡啶-3-基甲氧基)-7-((2-溴-[1,1'-联苯]-3-基)甲氧基)苯并[c][1,2,5]噁二唑-4-基)甲基)-d-丝氨酸(化合物13)
参照实施例12的方法,将实施例12中的l-丝氨酸乙酯盐酸盐替换成d-丝氨酸乙酯盐酸盐,制得化合物13:1hnmr(300mhz,cd3od)δ8.67(d,j=1.5hz,1h),8.51(dd,j=4.9,1.5hz,1h),8.10(d,j=7.6hz,1h),7.65(dd,j=7.6,1.6hz,1h),7.54–7.33(m,7h),6.96(s,1h),5.53(s,2h),5.40(s,2h),4.13(d,j=13.4hz,1h),4.06(d,j=13.6hz,1h),3.72(dd,j=10.7,5.6hz,1h),3.63(dd,j=10.7,6.2hz,1h),3.15(t,j=5.9hz,1h).hrms(esi):m/z629.08200[m+na]+。
实施例14
n-((5-((2-氰基吡啶-4-基)甲氧基)-7-((2-氯-[1,1'-联苯]-3-基)甲氧基)苯并[c][1,2,5]噁二唑-4-基)甲基)-d-丝氨酸(化合物14)
合成路线:
化合物c-2的合成
取化合物c-1(6.15g)溶于无水四氢呋喃(50ml)中,冰盐浴下缓慢滴加硼烷四氢呋喃复合物(60ml),室温下反应约5小时停止反应,冰浴下加水淬灭反应。用乙酸乙酯(50mlx4)萃取,和饱和食盐水(30mlx2)洗涤,无水硫酸钠干燥。减压蒸除溶剂,得白色固体化合物c-2(5.18g)。
化合物c-3的合成
取化合物c-2(2.5g),苯硼酸(1.6g),[1,1’-双(二苯基膦)二茂铁]二氯化钯二氯甲烷络合物(180mg),无水醋酸钾(2.1g),18-冠-6-醚(581mg)加入到100ml三颈瓶中,氩气保护,换气三次。室温下加入甲苯(21ml),无水乙醇(7ml)和水(3.5ml)。90℃油浴加热,反应约5小时后停止加热,减压蒸除溶剂,用乙酸乙酯25ml稀释,硅藻土抽滤,母液用乙酸乙酯(45mlx3)萃取,饱和食盐水(30mlx2)洗涤,无水硫酸钠干燥。减压蒸除溶剂,残余物经柱层析(洗脱剂:石油醚/乙酸乙酯=4:1)纯化,得无色油状化合物c-3(2.2g)。
化合物c-4的合成
取化合物c-3(2.2g),三苯基膦(5.3g),溶于无水四氢呋喃(30ml)中,于冰浴下缓慢加入四溴化碳(6.6g),室温下反应约4小时后停止反应。减压蒸除溶剂,用二氯甲烷(25ml)稀释后硅藻土抽滤,母液经柱层析(洗脱剂:石油醚/乙酸乙酯=5:1)纯化,得无色油状化合物c-4(2.4g)。
化合物iv-1的合成
取化合物iv-1(1.6g)溶于无水丙酮(25ml)中,加入无水碳酸氢钠(1.3g)和四丁基碘化胺(tbai,500mg),并缓慢加入c-4(2.03g),氩气保护,70℃油浴加热。反应约6小时后停止加热,减压蒸除溶剂,加入乙酸乙酯(15ml)稀释,用水(10mlx3)和饱和食盐水(10mlx2)洗涤,无水硫酸钠干燥。减压蒸除溶剂,残余物经柱层析(洗脱剂:石油醚/乙酸乙酯=5:1)纯化,得淡黄色固体化合物iv-1(960mg)。
化合物14的合成
参照实施例1的方法,将实施例1中的i-2替换成iv-1,3-羟甲基苯甲腈替换成2-氰基-4-羟甲基吡啶,制得化合物14:1hnmr(300mhz,cd3od)δ8.67(d,j=5.0hz,1h),8.01(s,1h),7.71(dd,j=5.4,1.0hz,1h),7.61(dd,j=7.1,2.3hz,1h),7.48–7.32(m,7h),6.85(s,1h),5.51(s,2h),5.45(s,2h),4.19(d,1h),4.15(d,j=13.2hz,1h),3.74(dd,1h),3.66(dd,j=10.9,6.3hz,1h),3.19(t,j=5.8hz,1h).ms(esi):m/z624.3[m+k]+。
实施例15
n-((5-((3-氰基苄基)氧基)-7-((2-氰基-[1,1'-联苯]-3-基)甲氧基)-苯并[c][1,2,5]噁二唑-4-基)甲基)-d-丝氨酸(化合物15)
合成路线:
化合物d-2的合成
取化合物d-1(10g)溶于甲醇(75ml)中,于室温下缓慢滴加浓硫酸(20ml),约15分钟加毕,85℃油浴加热。反应约15小时后停止加热,减压蒸除溶剂,用乙酸乙酯(60mlx4)萃取,饱和碳酸钠(50mlx3)和饱和食盐水(100mlx4)洗涤,无水硫酸钠干燥。减压蒸除溶剂,残余物经柱层析(洗脱剂:石油醚/乙酸乙酯=50:1)纯化,得淡黄色油状化合物d-2(9.0g)。
化合物d-3的合成
取化合物d-2(9.0g)溶于水(50ml)中,冰盐浴下缓慢加入盐酸(20ml),搅拌10分钟,缓慢滴加亚硝酸钠(3.2g)的水溶液,约30分钟加毕,冰盐浴下搅拌约45分钟后,向反应液中缓慢滴加碘化钾(8.4g)的水溶液,室温下反应8小时后停止反应。用硫代硫酸钠的饱和水溶液(60ml),乙酸乙酯(100mlx3)萃取,饱和食盐水(80mlx3)洗涤,无水硫酸钠干燥。减压蒸除溶剂,残余物经柱层析(洗脱剂:石油醚/乙酸乙酯=50:1)纯化,得淡黄色油状化合物d-3(11g)。
化合物d-4的合成
取化合物d-3(11g),氰化亚铜(11.5g),三(二亚苄基丙酮)二钯(604mg),双二苯基膦二茂铁(730mg),无水醋酸钾(6.5g),18-冠-6-醚(873mg)加入到100ml三颈瓶中,氩气保护,换气三次,加入无水1,4-二氧六环40ml,110℃油浴加热。反应约5小时后停止加热,减压蒸除溶剂,用乙酸乙酯(40ml)稀释,硅藻土抽滤,母液经柱层析(洗脱剂:石油醚/乙酸乙酯=3:1)纯化,得白色固体化合物d-4(3.47g)。
化合物d-5的合成
取化合物d-4(1.5g),苯硼酸(914mg),[1,1’-双(二苯基膦)二茂铁]二氯化钯二氯甲烷络合物(257mg),无水醋酸钾(1.2g),18-冠-6-醚(330mg)加入到50ml三颈瓶中,氩气保护,换气三次。室温下加入甲苯(18ml),无水乙醇(6ml),水(3ml)。90℃油浴加热。反应约5小时后停止加热,减压蒸除溶剂,用乙酸乙酯25ml稀释,硅藻土抽滤,母液用乙酸乙酯(40mlx3)萃取,饱和食盐水(40mlx3)洗涤,无水硫酸钠干燥。减压蒸除溶剂,残余物经柱层析(洗脱剂:石油醚/二氯甲烷=30:1)纯化,得无色油状化合物d-5(1.4g)。
化合物d-6的合成
取化合物d-5(1.41g)溶于甲醇(10ml)和四氢呋喃(10ml)的混合溶剂中,于冰浴下缓慢加入一水合氢氧化锂(619mg),室温下反应约6小时后停止反应,减压蒸除溶剂。1n盐酸酸化调ph=2~3,析出白色固体化合物d-6(1.36g)。
化合物d-7的合成
取化合物d-6(1.36g),溶于无水四氢呋喃(30ml)中,冰盐浴下缓慢滴加硼烷四氢呋喃复合物(12ml),室温下反应约5小时停止反应,冰浴下加水淬灭反应。用乙酸乙酯(30mlx3)萃取,和饱和食盐水(25mlx3)洗涤,无水硫酸钠干燥。减压蒸除溶剂,得白色固体化合物d-7(952mg)。
化合物d-8的合成
取化合物d-7(948mg),三苯基膦(1.78g),溶于无水四氢呋喃(30ml)中,于冰浴下缓慢加入四溴化碳(2.2g),室温下反应约4小时后停止反应。减压蒸除溶剂,用二氯甲烷(20ml)稀释,硅藻土抽滤,母液经柱层析(洗脱剂:石油醚/乙酸乙酯=5:1)纯化,得黄色油状化合物d-8(1.2g)。
化合物vi-1的合成
取化合物m-2(968mg)溶于无水丙酮(35ml)中,加入无水碳酸氢钠(750mg)和四丁基碘化胺(tbai,850mg),并缓慢加入d-8(1.18g),氩气保护,70℃油浴加热。反应约6小时后停止加热,减压蒸除溶剂,加入乙酸乙酯(35ml)稀释,用水(20mlx3)和饱和食盐水(15mlx2)洗涤,无水硫酸钠干燥。减压蒸除溶剂,残余物经柱层析(洗脱剂:石油醚/乙酸乙酯=3:1)纯化,得淡黄色固体化合物vi-1(1.38g)。
化合物15的合成
参照实施例1的方法,将实施例1中的i-2替换成vi-1,3-羟甲基苯甲腈替换成2-氰基-4-羟甲基吡啶,制得化合物15:1hnmr(300mhz,cd3od)δ8.69(d,j=5.1hz,1h),8.02(s,1h),7.84–7.70(m,3h),7.65–7.55(m,3h),7.56–7.45(m,3h),6.97(s,1h),5.62(s,2h),5.48(s,2h),4.21(d,j=13.5hz,1h),4.14(d,j=13.4hz,1h),3.75(dd,j=10.6,5.5hz,1h),3.66(dd,j=10.7,6.3hz,1h),3.19(t,j=5.9hz,1h).ms(esi):m/z615.4[m+k]+。
实施例16
n-((5-((3-氰基苄基)氧基)-7-((2-氰基-[1,1'-联苯]-3-基)甲氧基)-苯并[c][1,2,5]噁二唑-4-基)甲基)-d-丝氨酸(化合物16)
参照实施例15的方法,将实施例15中的2-氰基-4-羟甲基吡啶替换成3-羟甲基苯甲腈,制得化合物16:1hnmr(300mhz,cd3od)δ7.94–7.85(m,2h),7.79(d,j=2.8hz,1h),7.78(s,1h),7.69(d,j=7.7hz,1h),7.65–7.56(m,4h),7.56–7.47(m,3h),7.01(s,1h),5.63(s,2h),5.40(s,3h),4.16(d,j=13.5hz,1h),4.09(d,j=13.5hz,1h),3.73(dd,j=10.7,5.7hz,1h),3.64(dd,j=10.6,6.2hz,1h),3.16(t,j=5.7hz,1h).hrms(esi):m/z598.16974[m+na]+。
实施例17
n-((5-((2-氰基吡啶-4-基)甲氧基)-7-([1,1'-联苯]-3-基甲氧基)苯并[c][1,2,5]噁二唑-4-基)甲基)-d-丝氨酸(化合物17)
参照实施例1的方法,将实施例1中的i-1替换成3-苯基苄基溴,将3-羟甲基苯甲腈替换成2-氰基-4-羟甲基吡啶,制得化合物17:1hnmr(300mhz,dmso)δ8.65(d,j=5.0hz,1h),7.96(s,1h),7.74(s,1h),7.68–7.56(m,4h),7.49–7.40(m,4h),7.35(t,j=7.3hz,1h),6.84(s,1h),5.46(s,2h),5.40(s,2h),4.19(d,j=13.4hz,1h),4.12(d,j=13.4hz,1h),3.75(dd,j=10.7,5.5hz,1h),3.66(dd,j=10.6,6.2hz,1h),3.18(t,j=5.9hz,1h).ms(esi):m/z590.3[m+k]+。
实施例18
2-(((5-(吡啶-3-基甲氧基)-7-((2-溴-[1,1'-联苯]-3-基)甲氧基)苯并[c][1,2,5]噁二唑-4-基)甲基)氨基)乙-1-醇(化合物18)
参照实施例12的方法,将实施例12中的l-丝氨酸乙酯盐酸盐替换成乙醇胺,制得化合物18:ms(esi):m/z562.3[m+h]+。
实施例19
(s)-2-(((5-(吡啶-3-基甲氧基)-7-((2-溴-[1,1'-联苯]-3-基)甲氧基)苯并[c][1,2,5]噁二唑-4-基)甲基)氨基)-2-苯基乙酸(化合物19)
参照实施例12的方法,将实施例12中的l-丝氨酸乙酯盐酸盐替换成l-苯甘氨酸甲酯盐酸盐,制得化合物19:ms(esi):m/z652.3[m+h]+。
实施例20
(r)-2-(((5-(吡啶-3-基甲氧基)-7-((2-溴-[1,1'-联苯]-3-基)甲氧基)苯并[c][1,2,5]噁二唑-4-基)甲基)氨基)-2-苯基乙酸(化合物20)
参照实施例12的方法,将实施例12中的l-丝氨酸乙酯盐酸盐替换成d-苯甘氨酸甲酯盐酸盐,制得化合物20:ms(esi):m/z652.3[m+h]+。
实施例21
(2r,4r)-n-((5-(吡啶-3-基甲氧基)-7-((2-溴-[1,1'-联苯]-3-基)甲氧基)苯并[c][1,2,5]噁二唑-4-基)甲基)-4-羟基脯氨酸(化合物21)
参照实施例12的方法,将实施例12中的l-丝氨酸乙酯盐酸盐替换成(2r,4r)-4-羟基吡咯烷-2-羧酸甲酯盐酸盐,制得化合物21:ms(esi):m/z654.4[m+na]+。
实施例22
n-((5-((3-氰基苄基)氧基)-7-((2-甲基-[1,1'-联苯]-3-基)甲氧基)苯并[c][1,2,5]噁二唑-4-基)甲基)-d-丝氨酸乙酯(化合物22)
参照实施例1的方法制得化合物22(即实施例1中的i-7):ms(esi):m/z593.3[m+h]+。
实施例23
n-((5-((3-氰基苄基)氧基)-7-((3-(2,3-二氢苯并[b][1,4]二氧杂环己烯-6-基)-2-甲基苄基)氧基)苯并[c][1,2,5]噁二唑-4-基)甲基)-d-丝氨酸乙酯(化合物23)
参照实施例10的方法,不经过水解即可制得化合物23:ms(esi):m/z651.3[m+h]+。
实施例24
n-((5-((2-氰基吡啶-4-基)甲氧基)-7-((3-(2,3-二氢苯并[b][1,4]二氧杂环己烯-6-基)-2-甲基苄基)氧基)苯并[c][1,2,5]噁二唑-4-基)甲基)-d-丝氨酸乙酯(化合物24)
参照实施例10的方法,将实施例10中的3-羟甲基苯甲腈替换成2-氰基-4-羟甲基吡啶,且不经过水解即可制得化合物24:ms(esi):m/z674.6[m+na]+。
实施例25
n-((5-(吡啶-3-基甲氧基)-7-((2-溴-[1,1'-联苯]-3-基)甲氧基)苯并[c][1,2,5]噁二唑-4-基)甲基)-d-丝氨酸乙酯(化合物25)
参照实施例12的方法,将实施例12中的l-丝氨酸乙酯盐酸盐替换成d-丝氨酸乙酯盐酸盐,且不经过水解即可制得化合物25:ms(esi):m/z634.5[m+na]+。
实施例26
n-((5-(吡啶-3-基甲氧基)-7-((2-溴-[1,1'-联苯]-3-基)甲氧基)苯并[c][1,2,5]噁二唑-4-基)甲基)-d-丝氨酸乙酯盐酸盐(化合物26)
将实施例25中制得的化合物25加入盐酸乙醇溶液中,搅拌过夜,抽滤,制得化合物26,ms(esi):m/z656.5[m+na]+。
实施例27
n-((5-((2-氰基吡啶-4-基)甲氧基)-7-((2-氯-[1,1'-联苯]-3-基)甲氧基)苯并[c][1,2,5]噁二唑-4-基)甲基)-d-丝氨酸乙酯(化合物27)
参照实施例14的方法,不经过水解即可制得化合物27:ms(esi):m/z615.1[m+h]+。
实施例28
n-((5-((5-(甲基磺酰)吡啶-3-基)甲氧基)-7-((2-氯-[1,1’-联苯]-3-基)甲氧基)苯并[c][1,2,5]噁二唑-4-基)甲基)-d-丝氨酸(化合物28)
参照实施例14的方法,将实施例14中的2-氰基-4-羟甲基吡啶替换成(5-(甲基磺酰基)吡啶-3-基)甲醇,制得化合物28:ms(esi):m/z640.2[m+h]+。
实施例29
n-((5-((5-(甲基磺酰)吡啶-3-基)甲氧基)-7-((2-溴-[1,1’-联苯]-3-基)甲氧基)苯并[c][1,2,5]噁二唑-4-基)甲基)-d-丝氨酸(化合物29)
参照实施例13的方法,将实施例13中的3-吡啶甲醇替换成(5-(甲基磺酰基)吡啶-3-基)甲醇,制得化合物29:ms(esi):m/z684.5[m+h]+。
实施例30
n-((5-((2-氰基吡啶-4-基)甲氧基)-7-((2-溴-[1,1'-联苯]-3-基)甲氧基)苯并[c][1,2,5]噁二唑-4-基)甲基)-d-丝氨酸(化合物30)
参照实施例13的方法,将实施例13中的3-吡啶甲醇替换成2-氰基-4-羟甲基吡啶,制得化合物30:1hnmr(300mhz,methanol-d4)δ8.71(d,j=5.1hz,1h),8.27(s,1h),7.85(d,j=5.1hz,1h),7.61–7.53(m,1h),7.53–7.27(m,7h),6.94(s,1h),5.61(s,2h),5.54(s,2h),4.67(d,j=4.9hz,2h),4.03(dd,j=11.8,3.9hz,1h),3.86(dd,j=11.8,6.9hz,1h),3.73–3.62(m,1h).ms(esi):m/z631.5[m+h]+。
实施例31
n-((5-((5-氰基吡啶-3-基)甲氧基)-7-((2-溴-[1,1'-联苯]-3-基)甲氧基)苯并[c][1,2,5]噁二唑-4-基)甲基)-d-丝氨酸(化合物31)
参照实施例13的方法,将实施例13中的3-吡啶甲醇替换成5-(羟甲基)烟腈,制得化合物31:1hnmr(300mhz,methanol-d4)δ9.08(s,1h),8.93(s,1h),8.52(s,1h),7.64(d,j=7.7hz,1h),7.51–7.29(m,7h),7.00(s,1h),5.54(s,2h),5.49(s,2h),4.56–4.48(m,2h),4.27–4.20(m,2h),3.81–3.75(m,1h).ms(esi):m/z631.5[m+h]+。
实施例32
(s)-n-((5-((2-氰基吡啶-4-基)甲氧基)-7-((2-溴-[1,1'-联苯]-3-基)甲氧基)苯并[c][1,2,5]噁二唑-4-基)甲基)哌啶-2-羧酸(化合物32)
参照实施例30的方法,将实施例30中的d-丝氨酸乙酯盐酸盐替换成(s)-哌啶-2-甲酸甲酯盐酸盐,制得化合物32:ms(esi):m/z655.5[m+h]+。
实施例33
(s)-n-((5-(吡啶-3-基甲氧基)-7-((2-溴-[1,1'-联苯]-3-基)甲氧基)苯并[c][1,2,5]噁二唑-4-基)甲基)哌啶-2-羧酸(化合物33)
参照实施例12的方法,将实施例12中的l-丝氨酸乙酯盐酸盐替换成(s)-哌啶-2-甲酸甲酯盐酸盐,制得化合物33:ms(esi):m/z630.6[m+h]+。
实施例34
(s)-n-((5-((5-氰基吡啶-3-基)甲氧基)-7-((2-溴-[1,1'-联苯]-3-基)甲氧基)苯并[c][1,2,5]噁二唑-4-基)甲基)哌啶-2-羧酸(化合物34)
合成路线:
化合物e-1的合成
取化合物iii-1(3.5g)溶于乙腈(50ml)中,分批缓慢加入叠氮化钠(740mg),75℃油浴加热。反应约5小时后停止加热,减压蒸除溶剂,得黄色固体化合物e-1的粗品(3.9g)。
化合物e-2的合成
取化合物e-1的粗品(3.9g)溶于甲苯(30ml),置于110℃油浴加热。反应约5小时后停止加热,静置2小时,有固体析出,抽滤,得黄色固体化合物e-2(1.7g)。
化合物e-3的合成
取化合物e-2(900mg)加入乙腈(25ml)中,加入无水碳酸钾(655mg)和5-(羟甲基)烟腈(320mg),反应液在常温反应12小时后停止反应,减压蒸除溶剂,加入乙酸乙酯(25ml)稀释,用水(10mlx3)和饱和食盐水(10mlx2)洗涤,无水硫酸钠干燥。减压蒸除溶剂,残余物经柱层析(洗脱剂:石油醚/乙酸乙酯=1:1)纯化,得黄色固体化合物e-3(756mg)。
化合物e-4的合成
取化合物e-3(756mg)溶于n,n-二甲基甲酰胺(5ml),加入三苯基膦(365mg),90℃油浴加热。反应约6小时后停止加热,加入水(10ml),用乙酸乙酯(10mlx3)萃取,无水硫酸钠干燥。减压蒸除溶剂,残余物经柱层析(洗脱剂:石油醚/乙酸乙酯=2:1)纯化,得黄色固体化合物e-4(460mg)。
化合物e-5的合成
取化合物e-4(460mg)溶于无水乙醇(10ml)中,冰浴下加入硼氢化钠(28mg),反应约3小时后停止反应,加入水(10ml),用乙酸乙酯(10mlx3)萃取,无水硫酸钠干燥。减压蒸除溶剂,残余物经柱层析(洗脱剂:石油醚/乙酸乙酯=1:1)纯化,得黄色固体化合物e-5(420mg)。
化合物e-6的合成
取化合物e-5(420mg),三苯基膦(403mg),溶于无水四氢呋喃(10ml)中,于冰浴下缓慢加入四溴化碳(510mg),室温下反应约4小时后停止反应。减压蒸除溶剂,用二氯甲烷(10ml)稀释后硅藻土抽滤,母液经柱层析(洗脱剂:石油醚/乙酸乙酯=2:1)纯化,得淡黄色固体化合物e-6(233mg)。
化合物e-7的合成
取(s)-哌啶-2-甲酸甲酯盐酸盐(178mg)溶于n,n-二甲基甲酰胺(5ml),加入无水碳酸钾(91mg),室温搅拌20分钟,加入化合物e-6(200mg),70℃油浴加热。反应12小时后停止反应。减压蒸除溶剂,残余物经柱层析(洗脱剂:石油醚/乙酸乙酯=1:1)纯化,得黄色油状化合物e-7(135mg)。
化合物34的合成
取化合物e-7(135mg)溶于甲醇与四氢呋喃的混合溶剂(2:1,3ml),加入一水合氢氧化锂(25mg),室温反应5小时后,减压蒸除溶剂,残余物经柱层析(洗脱剂:甲醇/水=1:1)纯化,得淡黄色固体化合物34(70mg):ms(esi):m/z655.5[m+h]+。
实施例35
(s)-n-((5-((5-(甲基磺酰)吡啶-3-基)甲氧基)-7-((2-溴-[1,1’-联苯]-3-基)甲氧基)苯并[c][1,2,5]噁二唑-4-基)甲基)哌啶-2-羧酸(化合物35)
参照实施例34的方法,将实施例34中的5-(羟甲基)烟腈替换成(5-(甲基磺酰基)吡啶-3-基)甲醇,制得化合物35:ms(esi):m/z708.6[m+h]+。
实施例36
(s)-2-(((5-((2-氰基吡啶-4-基)甲氧基)-7-((2-溴-[1,1'-联苯]-3-基)甲氧基)苯并[c][1,2,5]噁二唑-4-基)甲基)氨基)-3-羟基-2-甲基丙酸(化合物36)
参照实施例30的方法,将实施例30中的d-丝氨酸乙酯盐酸盐替换成2-甲基-l-丝氨酸乙酯盐酸盐,制得化合物36:ms(esi):m/z645.5[m+h]+。
实施例37
(s)-2-(((5-((5-氰基吡啶-3-基)甲氧基)-7-((2-溴-[1,1'-联苯]-3-基)甲氧基)苯并[c][1,2,5]噁二唑-4-基)甲基)氨基)-3-羟基-2-甲基丙酸(化合物37)
参照实施例31的方法,将实施例31中的d-丝氨酸乙酯盐酸盐替换成2-甲基-l-丝氨酸乙酯盐酸盐,制得化合物37:ms(esi):m/z645.4[m+h]+。
实施例38
(s)-2-(((5-((5-氰基吡啶-3-基)甲氧基)-7-((2-溴-[1,1'-联苯]-3-基)甲氧基)苯并[c][1,2,5]噁二唑-4-基)甲基)氨基)-3-羟基-2-甲基丙酸(化合物38)
参照实施例12的方法,将实施例12中的l-丝氨酸乙酯盐酸盐替换成2-甲基-l-丝氨酸乙酯盐酸盐,制得化合物38:ms(esi):m/z620.5[m+h]+。
实施例39
(s)-2-(((5-((2-氰基吡啶-4-基)甲氧基)-7-((2-氯-[1,1'-联苯]-3-基)甲氧基)苯并[c][1,2,5]噁二唑-4-基)甲基)氨基)-3-羟基-2-甲基丙酸(化合物39)
参照实施例27的方法,将实施例27中的d-丝氨酸乙酯盐酸盐替换成2-甲基-l-丝氨酸乙酯盐酸盐,制得化合物39:ms(esi):m/z601.3[m+h]+。
实施例40
(s)-2-(((5-((5-氰基吡啶-3-基)甲氧基)-7-((2-氯-[1,1'-联苯]-3-基)甲氧基)苯并[c][1,2,5]噁二唑-4-基)甲基)氨基)-3-羟基-2-甲基丙酸(化合物40)
参照实施例14的方法,将实施例14中的2-氰基-4-羟甲基吡啶替换成3-吡啶甲醇,d-丝氨酸乙酯盐酸盐替换成2-甲基-l-丝氨酸乙酯盐酸盐,制得化合物40:ms(esi):m/z576.0[m+h]+。
实施例41
n-(2-(((5-(吡啶-3-基甲氧基)-7-((2-氯-[1,1'-联苯]-3-基)甲氧基)苯并[c][1,2,5]噁二唑-4-基)甲基)氨基)乙基)甲磺酰胺(化合物41)
参照实施例40的方法,将实施例40中的2-甲基-l-丝氨酸乙酯盐酸盐替换成甲磺酰基乙胺,制得化合物41:ms(esi):m/z595.1[m+h]+。
实施例42
2-(((5-(吡啶-3-基甲氧基)-7-((2-氯-[1,1'-联苯]-3-基)甲氧基)苯并[c][1,2,5]噁二唑-4-基)甲基)氨基)乙烷-1-磺酸(化合物42)
参照实施例40的方法,将实施例40中的2-甲基-l-丝氨酸乙酯盐酸盐替换成牛磺酸,制得化合物42:ms(esi):m/z582.4[m+h]+。
实施例43
n-(2-(((7-((2-氯-[1,1'-联苯]-3-基)甲氧基)-5-((5-(甲基磺酰基)吡啶-3-基)甲氧基)苯并并[c][1,2,5]噁二唑-4-基)甲基)氨基)乙基)甲磺酰胺(化合物43)
参照实施例41的方法,将实施例41中的3-吡啶甲醇替换成(5-(甲基磺酰基)吡啶-3-基)甲醇,制得化合物43:ms(esi):m/z673.2[m+h]+。
实施例44
n-((5-(吡啶-3-基甲氧基)-7-((2-氯-[1,1'-联苯]-3-基)甲氧基)苯并[c][1,2,5]噁二唑-4-基)甲基)-d-丝氨酸(化合物44)
参照实施例40的方法,将实施例40中的2-甲基-l-丝氨酸乙酯盐酸盐替换成d-丝氨酸乙酯盐酸盐,制得化合物44:ms(esi):m/z562.1[m+h]+。
实施例45
n-((5-((2-氰基吡啶-4-基)甲氧基)-7-((3-(2,3-二氢苯并[b][1,4]二氧杂环己烯-6-基)-2-溴苄基)氧基)苯并[c][1,2,5]噁二唑-4-基)甲基)-d-丝氨酸(化合物45)
参照实施例30的方法,将实施例30中的苯硼酸替换成苯并二氧六环-4-硼酸酯,制得化合物45:1hnmr(300mhz,methanol-d4)δ8.67(d,j=5.0hz,1h),8.00(s,1h),7.77–7.51(m,4h),7.42–7.36(m,1h),7.29(d,j=7.6hz,1h),6.89–6.78(m,2h),5.46(s,2h),5.44(d,j=7.3hz,2h),4.29(s,4h),4.22–4.05(m,2h),3.79–3.70(m,1h),3.70–3.61(m,1h),3.19(t,j=5.9hz,1h).ms(esi):m/z688.5[m+h]+。
实施例46
生物活性评价
1、本发明的化合物对pd-1/pd-l1蛋白-蛋白相互作用的抑制活性评价
实验目的:使用pd-1/pd-l1bindingassaykit检测试剂盒(cisbio公司),检测式(i)化合物抑制pd-1和pd-l1相互作用的活性。
实验原理:通过使用anti-tag1-europium(htrf供体)和anti-tag2-xl665(htrf受体)检测tag1-pd-l1和tag2-pd-1之间的相互作用。当htrf供体和htrf受体由于pd-l1和pd-1结合而紧密接近时,htrf供体的激发触发朝向htrf受体的荧光共振能量转移(fret),htrf受体又在665nm处发射荧光。该特定信号与pd-1/pd-l1相互作用的程度成正比。因此,阻断pd-1/pd-l1相互作用的化合物或抗体将导致htrf信号的减少。
实验材料:384孔细胞培养板购自nunc公司;均相时间分辨荧光homogeneoustime-resolvedfluorescence(htrf)试剂盒购自cisbio公司;dmso购自sigma公司。
实验仪器:perkinelmerenvision多功能酶标检测仪。
受试药物:式(i)化合物。样品用dmso配制成母液使用,使用时用diluentbuffer稀释使用,dmso终浓度不超过0.1%。
实验方案:pd-1/pd-l1蛋白-蛋白相互作用抑制试验采用htrf试剂盒。设立阴性对照组、阳性对照组和给药组(包括阳性药和受试化合物),每组3个复孔。在阴性对照组中,向384孔板中依次加入2μldiluentbuffer、4μl经diluentbuffer稀释的tag1-pd-l1和4μl经diluentbuffer稀释的tag2-pd-1;在阳性对照组中,向384孔板中依次加入2μldiluentbuffer、4μl经diluentbuffer稀释的tag1-pd-l1和4μldiluentbuffer;在给药组中,向384孔板中依次加入2μl经diluentbuffer稀释的受试药物[阳性药或式(i)化合物],随后每孔各加入4μl经diluentbuffer稀释的tag1-pd-l1和4μl经diluentbuffer稀释的tag2-pd-1。待各组加完后,将384孔板于37℃下孵育15分钟后,再向每孔加入5μl经detectionbuffer稀释的anti-tag1-eu3+和anti-tag2-xl665。于37℃下孵育1小时至过夜后,用perkinelmerenvision多功能酶标检测仪测定在665nm和620nm处的荧光值,htrfratio=(665nm/620nm)*104。每个化合物检测8-10个浓度梯度,使用graphpad软件计算化合物的ic50。本实验选用bms公司专利wo2015160641中的化合物1016(bms-1016)为阳性药。活性测试结果见表2。
表2、化合物抑制pd-1/pd-l1蛋白-蛋白相互作用的蛋白水平活性
实验结果表明,本发明的化合物具有显著的pd-1/pd-l1蛋白-蛋白相互作用的抑制活性。例如,化合物11(ic50=2.719nm)、化合物13(ic50=2.061nm)和化合物14(ic50=1.787nm)等的活性显著优于阳性对照bms-1016(ic50=32.20nm)。本发明中的其他化合物等也都显示了显著的pd-1/pd-l1抑制活性,例如,化合物28、29、30、31、32、33、34、35、37、38、39、41、43、44和45等的ic50值均在10nm以下。这提示本发明的苯并噁二唑类化合物可用作免疫检查点pd-1/pd-l1抑制剂。
2、本发明的化合物阻断hpd-l1蛋白抑制人外周血单个核细胞(pbmc)分泌ifn-γ作用的实验
人外周血单个核细胞(pbmc)活化后会分泌ifn-γ、il-2和tnf-α等细胞因子,而hpd-l1(人源pd-l1)蛋白与人pbmc上的pd-1蛋白结合会抑制pbmc的活化,降低细胞因子分泌。本实验的目的是检测化合物阻断hpd-l1蛋白抑制pbmc活化的能力。
具体操作如下:采用澳赛尔斯生物技术有限公司购得的人pbmc,接种到96孔板中,每孔接种1ⅹ105个细胞。实验共分五组,一组为空白对照(只加入pbmc),一组为激活组(pbmc+anti-cd3/anti-cd28抗体),一组为抑制组(pbmc+anti-cd3/anti-cd28抗体+hpd-l1蛋白),其他两组分别为在抑制组的基础上加入1μm和10nm的化合物12。48小时后采用biolegend公司的ifn-γelisa检测试剂盒检测细胞上清液中ifn-γ的含量。实验结果显示,激活组中pbmc受到anti-cd3/anti-cd28抗体作用后,分泌ifn-γ显著增加,说明pbmc呈激活状态;抑制组中hpd-l1蛋白抑制pbmc分泌ifn-γ,说明pbmc的激活状态受到抑制;化合物12具有显著的阻断pd-l1抑制人pbmc细胞分泌ifn-γ的作用(图1)。本发明中的其他化合物等在该实验中也表现出相似的效果,例如,化合物9、10、11、13、14、15、16、28、29、30、31、32、33、34、35、37、38、39、41、43、44和45等在1μm和10nm浓度下具有显著的阻断pd-l1抑制人pbmc细胞分泌ifn-γ的作用。这表明本发明的化合物具有抑制pd-1/pd-l1相互作用进而增强t细胞免疫活性的功效。
3、本发明的化合物阻断肿瘤细胞抑制人t细胞增殖作用的实验
人乳腺癌细胞mda-mb-231表面表达有pd-l1蛋白,当激活的人t细胞与mda-mb-231细胞接触后,会激活pd-1/pd-l1通路,进而抑制t细胞的激活,引起t细胞增殖变缓,并减少ifn-γ、il-2和tnf-α等细胞因子的释放。本实验的目的是检测化合物阻断mda-mb-231细胞抑制人t细胞增殖的能力。
具体操作如下:将对数期生长的mda-mb-231细胞接种到96孔板中,待生长到合适细胞密度,将分选得到的人t细胞悬液加入上清中,用anti-cd3/anti-cd28抗体激活t细胞,加入待测化合物。48小时后通过流式细胞仪检测t细胞的增殖情况。实验共分六组,一组为空白对照组(只加入t细胞),一组为激活组(t细胞+anti-cd3/anti-cd28抗体),一组为抑制组(t细胞+anti-cd3/anti-cd28抗体和mda-mb-231),其他三组分别为在抑制组的基础上加入10μm、100nm和10nm的化合物12。实验结果显示,激活组中t细胞受到anti-cd3/anti-cd28抗体作用后,增殖显著增加,说明t细胞呈激活状态;抑制组中人乳腺癌细胞mda-mb-231与t细胞共培养后,t细胞增殖能力下降,说明t细胞的激活状态受到抑制;化合物12具有显著的阻断肿瘤细胞抑制人t细胞增殖的能力(图2)。本发明中的其他化合物等在该实验中也表现出相似的效果,例如,化合物11、13、14、16、28、29、31、34、35、37、43、44和45等在10μm、100nm和10nm浓度下具有显著的阻断肿瘤细胞抑制人t细胞增殖的能力。这表明本发明的化合物可促进t细胞的增殖,增强t细胞的免疫功能,进而可抑制pd-1/pd-l1介导的肿瘤细胞免疫逃逸。
4、本发明的化合物阻断肿瘤细胞抑制人t细胞分泌ifn-γ作用的实验
如前所述,人乳腺癌细胞mda-mb-231表面表达有pd-l1蛋白,当激活的人t细胞与mda-mb-231细胞接触后,会激活pd-1/pd-l1通路,进而减少ifn-γ、il-2和tnf-α等细胞因子的释放。本实验的目的是检测化合物阻断mda-mb-231细胞抑制人t细胞分泌ifn-γ的能力。
具体操作如下:将对数期生长的mda-mb-231细胞接种到96孔板中,待生长到合适细胞密度,将分选得到的人t细胞悬液加入上清中,用anti-cd3/anti-cd28抗体激活t细胞,加入待测化合物。48小时后采用biolegend公司的ifn-γelisa检测试剂盒检测细胞上清液中ifn-γ的含量。实验共分六组,一组为空白对照组(只加入t细胞),一组为激活组(t细胞+anti-cd3/anti-cd28抗体),一组为抑制组(t细胞+anti-cd3/anti-cd28抗体+mda-mb-231),其他三组分别为在抑制组的基础上加入10μm、100nm和10nm的化合物12。实验结果显示,激活组中t细胞受到anti-cd3/anti-cd28抗体作用后,分泌ifn-γ显著增加,说明t细胞呈激活状态;抑制组中人乳腺癌细胞mda-mb-231与t细胞共培养后,t细胞分泌ifn-γ的水平下降,说明t细胞的激活状态受到抑制;化合物12具有显著的阻断肿瘤细胞抑制人t细胞分泌ifn-γ的能力(图3)。本发明中的其他化合物等在该实验中也表现出相似的效果,例如,化合物11、13、14、16、28、29、31、34、35、37、43、44和45等在10μm、100nm和10nm的浓度下具有显著的阻断肿瘤细胞抑制人t细胞分泌ifn-γ作用的能力。这表明本发明的化合物可阻断pd-1/pd-l1对t细胞的免疫抑制作用,恢复活化的t细胞分泌ifn-γ等细胞因子的能力,增强t细胞的免疫活性,进而增强t细胞的抗肿瘤免疫活性。
实施例47
片剂
将实施例14中制得的化合物14(50g)、羟丙甲基纤维素e(150g)、淀粉(200g)、聚维酮k30适量和硬脂酸镁(1g)混合,制粒,压片。
此外,可以根据药典2015版常规制剂法,将实施例1~45制得的化合物赋予不同的药物辅料制成胶囊剂、散剂、颗粒剂、丸剂、注射剂、糖浆剂、口服液、吸入剂、软膏剂、栓剂或贴剂等。
- 还没有人留言评论。精彩留言会获得点赞!