泊沙康唑的制备方法与流程
本发明涉及制药领域,更具体地涉及一种泊沙康唑的制备方法。
背景技术:
泊沙康唑(posaconazole)化学名为4-[4-[4-[4-[[(3r,5r)-5-(2,4-二氟苯基)-5-(1,2,4-三唑-1-基甲基)氧杂戊环-3-基]甲氧基]苯基]哌嗪-1-基]苯基-2-[(2s,3s)-2-羟基戊-3-基]-1,2,4-三唑-3-酮,是二代新型三唑类药物,由schering-plough研发,2005年首次在德国上市,商品名为noxafil。随后在澳大利亚,英国,美国,欧盟等多个国家上市,用于预防和治疗侵袭性真菌感染、耐药或其他药物无效的侵袭性真菌感染。泊沙康唑的结构式如下:
国内外已经有大量文献报道泊沙康唑的合成,化合物i是用于制备泊沙康唑的一种关键中间体,通过在适当的反应条件下将化合物i脱苄基可制备泊沙康唑。
目前化合物i脱苄基制备式a所示的泊沙康唑的方法主要有以下几种:①无机质子酸脱苄基,如浓盐酸、氢溴酸等;②三氯化硼脱苄基;③金属催化剂和氢气脱苄基。
专利cn102863431a公开:化合物i和盐酸进行反应,反应液用水稀释,碱性水溶液调ph值至8-11,萃取,有机相用氢氧化钠水溶液洗涤,制得泊沙康唑粗品。
专利cn105985329a公开:将化合物i溶于有机溶剂中,在氮气保护下降温至-80~-40℃,然后加入三氯化硼,升温至-10~-30℃反应;反应完毕用碱调节ph值至9-11,分离有机相,干燥得到泊沙康唑。
专利cn106366076a公开:以氢气为氢源,在氢化脱苄基反应条件下,将化合物i与催化剂和酸性试剂在反应溶剂中搅拌接触反应,得到混合溶液,过滤;滤液降温至0-5℃,滴加氢氧化钠溶液调节ph值至10-12,析出固体,过滤得到泊沙康唑。
专利cn108239077a公开:在甲酸和钯碳作用下,化合物i进行脱保护基和酯化反应生成化合物ii;在水性溶剂中,化合物ii在碱溶液条件下经酯水解反应,生成泊沙康唑。
现已公开的经化合物i脱苄基制备泊沙康唑的各种方法,其主要存在以下问题:
(1)采用无机强酸如盐酸、氢溴酸脱苄基,反应一般需要加热,化合物i较难反应完全,由于化合物i和泊沙康唑的溶解性相差不大,纯化困难,难以获得很高纯度的泊沙康唑;且热酸液对设备腐蚀性较强,因此对设备的耐酸碱要求较高;后处理酸液,产生大量三废;浓盐酸或氢溴酸等脱苄还会产生卤代的基因毒性杂质。
(2)采用三氯化硼脱苄基,反应往往需要在低温下进行,工业化生产难以实现;且三氯化硼沸点低易潮解,运输投料均有较大安全风险,不便于操作;同时与浓盐酸或氢溴酸类似,该法也会产生大量三废,还会产生氯代的基因毒性杂质。
(3)以氢气作氢源,钯催化氢化反应脱苄基,反应通常需要耐高压装置,且一般在酸性条件下进行,对反应设备的要求很高,操作繁琐复杂,生产时有易燃易爆的危险,存在较大的安全隐患。
(4)以甲酸作氢源,钯碳作催化剂,转移催化氢化脱苄基,先生成泊沙康唑甲酸酯的中间体,再经碱液进行酯水解反应才能生成泊沙康唑,后处理较为繁琐,且杂质较多不易除去。
目前,急需要提供一种操作简单、安全系数较高,纯度高、质量可控,且对环境友好,适宜于工业化生产的泊沙康唑制备方法。
技术实现要素:
本发明的目的在于克服现有技术中通过化合物i制备泊沙康唑的制备方法收率低,纯度低,潜在的基因毒性或者采用酸性试剂腐蚀性强以及后处理繁琐等缺陷,提供一种与现有技术不同的泊沙康唑的制备方法。本发明的制备方法,条件温和,得到的产品收率高、纯度高,而且避免了使用酸性试剂,省去用碱液调ph值的操作,从而简化了后处理,节约成本,避免大量废水的产生,整个过程安全可控,适合大规模工业化生产。
本发明是通过以下技术方案解决上述技术问题。
本发明提供了一种泊沙康唑的制备方法,其包括以下步骤:在甲酸铵和催化剂存在的条件下,在有机溶剂中,将化合物i进行如下所示的脱苄反应,得到化合物a即可;
所述的脱苄反应的操作和条件可为本领域常规的操作和条件,本发明优选以下操作和条件:
其中,所述甲酸铵与所述化合物i的摩尔比可为≥5:1,优选5:1~15:1,更优选8:1~12:1,例如10:1。
其中,所述有机溶剂可为醇类溶剂、醚类溶剂和酯类溶剂中的一种或多种,优选醇类溶剂。当所述有机溶剂为醇类溶剂时,所述醇类溶剂可为甲醇、乙醇、正丙醇和异丙醇中的一种或多种,优选甲醇。当所述有机溶剂为醚类溶剂时,所述醚类溶剂可为四氢呋喃和/或2-甲基四氢呋喃,优选四氢呋喃。当所述有机溶剂为酯类溶剂时,所述酯类溶剂可为乙酸乙酯。
其中,所述有机溶剂的体积与所述化合物i的质量比可为10~40ml/g,优选10~20ml/g,例如10ml/g。
其中,所述催化剂可为本领域常规的催化剂,优选湿钯碳、干钯碳、氢氧化钯、金属铂或铂碳,更优选湿钯碳或氢氧化钯。所述湿钯碳可为本领域常规的湿钯碳,例如10%的湿钯碳,优选含水量为0.5~65%,其中所述“%”为各组分的质量占各组分总质量的百分比。所述干钯碳可为本领域常规的干钯碳,例如10%的干钯碳,其中所述“%”为各组分的质量占各组分总质量的百分比。
其中,所述催化剂的用量可为本领域常规的用量,优选所述催化剂的用量为所述化合物i的重量的5~30%,优选14~25%,例如15%,16%,20%,所述“%”为所述催化剂的质量占所述化合物i的质量的重量百分比。
其中,所述脱苄反应的温度可为本领域常规的温度,例如30℃至溶剂的沸点。所述脱苄反应的温度优选30~80℃,更优选50~75℃,更进一步优选60~75℃,最优选60~65℃。
其中,所述脱苄反应的进程可采用本领域常规的监测方法进行监测(例如hplc、lc-ms、tlc),一般以所述化合物i消失或者不再变化作为反应的终点。所述脱苄反应的时间可为10~18h,例如16~18h。
本发明中,所述脱苄反应结束后,还可进一步包括以下后处理步骤,将所述反应液过滤、浓缩、溶解、水洗和浓缩,得到化合物a泊沙康唑粗品。
其中,所述过滤的操作和条件可为本领域常规的操作和条件,优选热过滤。所述热过滤的温度优选30-60℃,更优选40~50℃。
其中,所述溶解所用的溶剂可为卤代烃类溶剂和/或酯类溶剂。所述卤代烃类溶剂可为二氯甲烷和/或氯仿,优选二氯甲烷。所述酯类溶剂可为乙酸乙酯、甲酸乙酯、乙酸甲酯和乙酸异丙酯中的一种或多种,优选乙酸乙酯。
其中,所述水洗的操作和条件可为本领域常规的操作和条件,优选采用盐水洗涤。
其中,所述浓缩的操作和条件可为本领域常规的操作和条件。所述浓缩操作步骤前可进一步优选干燥。
其中,所述化合物a泊沙康唑粗品还可进一步采用重结晶纯化的方法得到泊沙康唑纯品;所述重结晶的溶剂为良溶剂和醇类溶剂的混合溶剂,所述良溶剂为酮类溶剂、酯类溶剂和卤代烃类溶剂中一种或多种;所述良溶剂和醇类溶剂的体积比为1:(5~12)。所述良溶剂和醇类溶剂的体积比优选为1:(8~12),更优选为1:(8-11)。所述良溶剂优选酮类溶剂。所述醇类溶剂可为甲醇、乙醇、正丙醇和异丙醇中的一种或多种,优选甲醇。所述酮类溶剂优选丙酮和/或甲乙酮,优选丙酮。所述酯类溶剂优选乙酸乙酯。所述卤代烃类溶剂优选二氯甲烷。
本发明中,“eq”是指“当量”,为化学专业用语,其指该物质与特定物质的摩尔数相当的量。
在不违背本领域常识的基础上,上述各优选条件,可任意组合,即得本发明各较佳实例。
本发明中化合物i可以参照专利cn102863431a中实施例2的制备方法制得,其它所用试剂和原料均市售可得。
本发明的积极进步效果在于:
1、本发明反应过程和后处理简单,所得产品收率高,纯度高,且避免了卤代的基因毒性杂质的产生,保证产品质量。
2、本发明的泊沙康唑的制备方法,避免使用酸性试剂,省去用碱液调ph值的操作,操作简便,节约成本的同时避免大量废水的产生,对环境友好。
3、本发明的制备方法在常压条件下即可进行反应,对设备要求较低,大大降低了生产成本,而且反应温度和反应条件要求不苛刻,过程安全可控,适合工业化生产。
附图说明
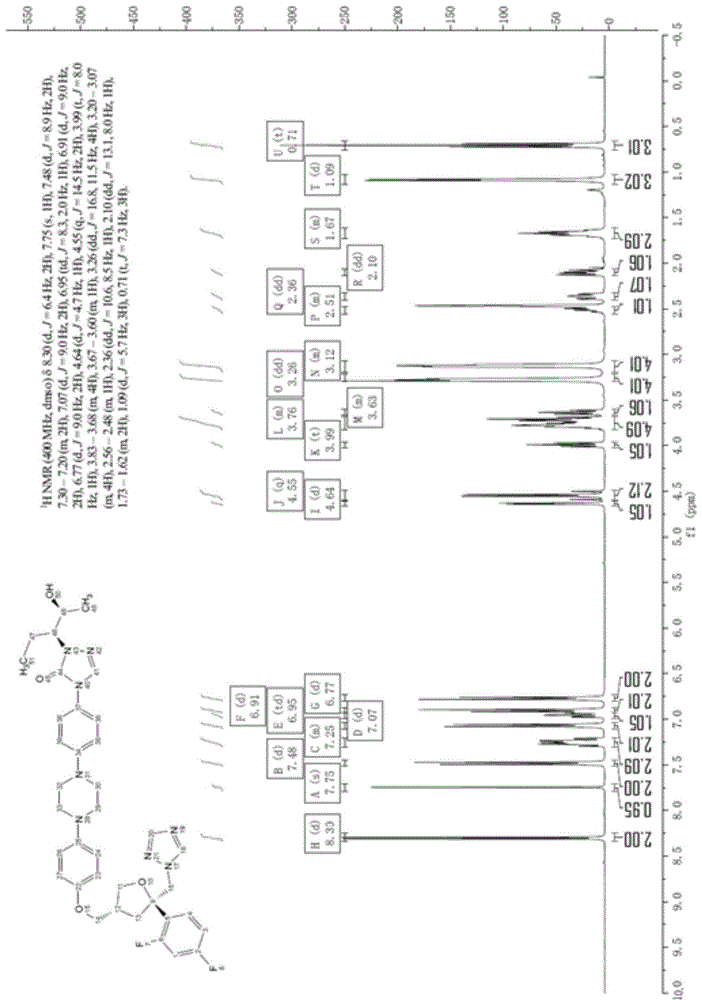
图1为实施例1得到泊沙康唑的核磁图谱。
具体实施方式
下面通过实施例的方式进一步说明本发明,但并不因此将本发明限制在所述的实施例范围之中。下列实施例中未注明具体条件的实验方法,按照常规方法和条件,或按照商品说明书选择。
实施例1
将5g式i化合物加入到50ml甲醇中,再加入1g湿钯碳(10%钯碳,水的含量为61%),4g甲酸铵(10eq),在60-65℃反应16-18h,至原料反应完全,hplc检测反应液中泊沙康唑的纯度98.00%。
将反应液在40-50℃下热过滤,滤液浓缩至干,将所得到的固体溶于二氯甲烷中,有机相用盐水洗,浓缩得到粗品3.9g,收率为88.04%,hplc检测纯度97.94%。
将所得粗品用39ml甲醇和3.9ml丙酮混合液重结晶,得到泊沙康唑纯品3.3g,收率为74.50%,经hplc检测纯度为99.77%。
1hnmr(400mhz,dmso):δ8.30(d,j=6.4hz,2h),7.75(s,1h),7.48(d,j=8.9hz,2h),7.30-7.20(m,2h),7.07(d,j=9.0hz,2h),6.95(td,j=8.3,2.0hz,1h),6.91(d,j=9.0hz,2h),6.77(d,j=9.0hz,2h),4.64(d,j=4.7hz,1h),4.55(q,j=14.5hz,2h),3.99(t,j=8.0hz,1h),3.83-3.68(m,4h),3.67-3.60(m,1h),3.26(dd,j=16.8,11.5hz,4h),3.20-3.07(m,4h),2.56-2.48(m,1h),2.36(dd,j=10.6,8.5hz,1h),2.10(dd,j=13.1,8.0hz,1h),1.73-1.62(m,2h),1.09(d,j=5.7hz,3h),0.71(t,j=7.3hz,3h);其核磁图谱如图1所示。
实施例2
将5g式i化合物加入到50ml甲醇中,再加入0.8g湿钯碳(10%钯碳,水的含量为61%),2g甲酸铵(5eq),60~65℃反应20h,hplc检测反应液中泊沙康唑的纯度为95.5%。
后处理同实施例1,得到粗品3.7g,收率为83.53%,hplc检测纯度98.32%。
得到泊沙康唑纯品3.1g,收率为67.72%,经hplc检测纯度为99.75%。其核磁数据基本同实施例1。
实施例3
将5g式i化合物加入到80ml异丙醇中,再加入1g氢氧化钯,4.8g甲酸铵(12eq),70-75℃反应至原料反应完全,hplc检测反应液中泊沙康唑的纯度为96.8%。
反应液在40-50℃下热过滤,滤液浓缩至干,将所得到的固体溶于乙酸乙酯中,有机相用盐水洗,浓缩得到粗品4.0g,收率为90.23%,hplc检测纯度98.63%。
将所得粗品用32ml甲醇和8ml丙酮混合液加热至溶清,再缓慢降温至室温,过滤,得到泊沙康唑3.43g,收率为77.42%,经hplc检测纯度为99.68%。其核磁数据基本同实施例1。
实施例4
参考实施例1的制备方法,将5g式i化合物加入到50ml甲醇中,再加入0.7g湿钯碳(10%钯碳,水的含量为61%),3.2g甲酸铵(8eq),60-65℃反应至原料反应完全,hplc检测反应液中泊沙康唑的纯度为97.5%。
将反应液在40-50℃下热过滤,滤液浓缩至干,将所得到的固体溶于二氯甲烷中,有机相用盐水洗,浓缩得到粗品3.8g,收率为85.77%,hplc检测纯度97.75%。
将所得粗品用60ml甲醇加热至溶清,再缓慢降温至室温,过滤,得到泊沙康唑3.35g,收率为75.62%,经hplc检测纯度为99.65%。其核磁数据基本同实施例1。
实施例5
将5g式i化合物加入到60ml乙醇中,再加入1g湿钯碳,6g甲酸铵(15eq),65~70℃反应至原料反应完全,hplc检测反应液中泊沙康唑的纯度为97.8%。
将反应液在40-50℃下热过滤,滤液浓缩至干,将所得到的固体溶于二氯甲烷中,有机相用盐水洗,浓缩得到粗品3.9g,收率为88.03%,hplc检测纯度97.86%。
将所得粗品用60ml乙醇加热至溶清,再缓慢降温至室温,过滤,得到泊沙康唑3.41g,收率为76.97%,经hplc检测纯度为99.59%。其核磁数据基本同实施例1。
实施例6
将实施例1中甲酸铵的用量4g甲酸铵(10eq),替换为4.5g(12eq),按照实施例1的条件进行反应,经hplc检测,反应液中,无原料剩余,泊沙康唑的含量为98.03%。
按照实施1的后处理方法,得到泊沙康唑粗品4.0g,收率为90.23%,hplc检测纯度96.52%。
得到纯品3.3g,收率为74.49%,经hplc检测纯度为99.76%。其核磁数据基本同实施例1。
实施例7
将实施例1中甲酸铵的用量4g甲酸铵(10eq),替换为6g(15eq),按照实施例1的条件进行反应,经hplc检测,反应液中,无原料剩余,泊沙康唑的含量为98.00%。
按照实施1的后处理方法,得到泊沙康唑粗品3.8g,收率为85.77%,hplc检测纯度98.53%。
得到纯品3.3g,收率为74.48%,经hplc检测纯度为99.82%。其核磁数据基本同实施例1。
实施例8
将实施例1中甲酸铵的用量4g甲酸铵(10eq),替换为2g(5eq),按照实施例1的条件进行反应,经hplc检测,反应液中,无原料剩余,泊沙康唑的含量为97.39%。
按照实施1的后处理方法,得到泊沙康唑粗品3.7g,收率为83.53%,hplc检测纯度97.82%。
得到纯品3.2g,收率为72.24%,经hplc检测纯度为99.58%。其核磁数据基本同实施例1。
实施例9
将实施例1中甲酸铵的用量4g甲酸铵(10eq)替换为1.6g(4eq),按照实施例1的条件进行反应,经hplc检测,反应液中,式i化合物的含量为60.3%(原料剩余),泊沙康唑的含量为35.86%。
延长反应时间至25小时,经hplc检测,反应液中,式i化合物的含量无明显下降。
按照实施1的后处理方法,得到泊沙康唑粗品4.2g,收率为94.80%,hplc检测纯度41.02%。纯品1.3g,收率为29.34%,经hplc检测纯度为80.22%。
实施例10
将实施例1中的溶剂甲醇替换为乙酸乙酯,按照实施例1的条件进行反应,hplc检测反应液中泊沙康唑的纯度为85.06%。
按照实施1的后处理方法,得到泊沙康唑粗品3.8g,收率为85.77%,hplc检测纯度86.02%。
得到纯品2.3g,收率为51.9%,经hplc检测纯度为98.36%。其核磁数据基本同实施例1。
实施例11
将实施例1中的溶剂甲醇替换为四氢呋喃,按照实施例1的条件进行反应,hplc检测反应液中泊沙康唑的纯度为86.63%。
按照实施1的后处理方法,得到泊沙康唑粗品4.1g,收率为92.54%,hplc检测纯度90.05%。
得到纯品2.6g,收率为56.43%,经hplc检测纯度为97.58%。其核磁数据基本同实施例1。
实施例12
将实施例1中的溶剂甲醇替换为乙腈,按照实施例1的条件进行反应,经hplc检测,反应液中,式i化合物的含量为72.56%,泊沙康唑的含量为19.02%。
延长反应时间至48小时,经hplc检测,反应液中,式i化合物的含量为70.85%,泊沙康唑的含量为19.12%。
按照实施1的后处理方法,得到泊沙康唑粗品4g,收率为90.29%,hplc检测纯度18.05%。纯品0.8g,收率为18.06%,经hplc检测纯度为53.71%。
对比例1
将5g式i化合物加入到30ml浓盐酸中,40~60℃保温反应10h,hplc检测反应液中原料含量2.3%。反应液降至室温,加入适量水稀释,用二氯甲烷萃取,水相用碱液调ph值至7~8,再用二氯甲烷萃取,浓缩得到粗品3.4g。所得粗品用甲醇和丙酮混合液结晶,过滤得到泊沙康唑2.9g,收率为65.46%,hplc检测纯度99.70%。
对比例2
将5g式i化合物加入到30ml甲酸中,1g湿钯碳,50~60℃反应至原料消失。过滤浓缩,加入甲醇,用氨水调ph至8~9,过滤得到泊沙康唑甲酸酯。将其加入到甲醇中,加入适量氨水并回流至泊沙康唑甲酸酯水解完全。加入纯化水,降温至室温过滤得到泊沙康唑粗品3.4g。所得粗品用甲醇和丙酮混合液结晶,过滤得到泊沙康唑3.0g,收率为67.72%,纯度99.51%。
对比例3
将5g式i化合物加入到50ml甲醇中,再加入1g湿钯碳,2.9g甲酸(10eq),60-65℃反应20h。经lc-ms检测反应液,原料含量67.74%,泊沙康唑含量23.63%,延长反应时间,原料仍然无法反应完全。
发明人提高甲酸用量至20eq、50eq、100eq,等比例提高溶剂用量,进行对比实验,经lc-ms检测原料转化率有所提升,但仍有超过30%的原料无法反应完全。
对比例1中,虽然得到的泊沙康唑产品的纯度与本发明相当,但是其使用了含卤强酸,醇羟基在浓盐酸或浓溴酸中加热会产生相应的卤代物,产生多个含基毒警示结构的杂质(参考专利cn109053708a)。故该方法得到的产品有着潜在的卤代的基因毒杂质,而且强酸还会腐蚀设备,后处理还需要调节ph值,产生大量的三废。
对比例2中,当以甲酸为溶剂和供氢体时,需要两步才能得到泊沙康唑,且产品的纯度偏低。
对比例3中,当以甲酸为供氢体时,仍有超过30%的原料无法反应完全,反应收率较低。
应当理解,本文所述的实施例仅用于说明目的,通过实施例将有助于进一步理解本发明,但不用于限制本发明的内容。对于本领域技术人员而言,对于原料和方法两者的许多改变可在不脱离本发明范围的情况下实施,这些改变或改进包括在本申请的范围内。
- 还没有人留言评论。精彩留言会获得点赞!