用于沉默嗜热毁丝霉中腺苷酸环化酶基因的干扰表达载体的制作方法
本发明属于微生物技术领域,尤其涉及用于沉默嗜热毁丝霉中腺苷酸环化酶基因的干扰表达载体。
背景技术:
随着工业机械化的不断发展以及汽车等耗能设备的大规模使用使得人们对化石等不可再生能源的需求越来越大。但是不可再生能源的数量有限且日渐枯竭,人们将目光转向于自然界中丰富的可再生资源,来缓解目前的能源压力。
木质纤维素是世界上规模最大、分布范围最广、含量最丰富的一类可再生资源,主要以草木、植物落叶、农业废弃物、木材处理废料、食品加工废料、市政废物等形式存在。木质纤维素相对廉价,如果能够对其进行高效利用将其转化为各种为人类所需的能源物质,将有助于缓解目前的能源压力,减少燃烧农作物秸秆所造成的环境污染,起到一定的环境保护作用。
纤维素酶是一类多组分复杂酶系。一般分为3大类,分别是内切葡聚糖酶(eg),外切葡聚糖酶(cbh)和β-葡萄糖苷酶(bg)。三种酶通过协同作用来降解大分子纤维素产生可利用的单糖从而转化为其他能源物质。eg可以将纤维素非结晶区内部的糖苷键随机水解,产生纤维糊精和低聚糖等物质。cbh能够将纤维素链的还原端或非还原端水解,并释放出纤维二糖。bg将纤维二糖和寡糖水解,并释放可以发酵的葡萄糖。
大自然中,真菌、细菌等微生物都能产生纤维素酶,但是一般应用于工业生产中的多是丝状真菌。经研究发现,与细菌产纤维素酶相比,丝状真菌产酶能力更强,所产的酶系更全、活性更高并且能够分泌到胞外。目前,能够大量分泌纤维素酶的丝状真菌有里氏木霉(trichodermareesei)、草酸青霉(penicilliumoxalicum)、黑曲霉(aspergillusniger)、粗糙脉孢菌(neurosporacrassa)等,其中里氏木霉被广泛应用于工业化生产。
嗜热毁丝霉(myceliophthorathermophila)是一类耐高温的嗜热丝状子囊真菌,其最低生长温度大于等于20℃,最高生长温度可大于等于50℃。它可以产生耐高温的纤维素酶,所产生的大多纤维素酶最适温度在60-80℃之间,所以被称为生产热稳定性纤维素酶的酶工厂,也被称为潜在的高效中高温酶库。
然而,嗜热毁丝霉纤维素酶基因表达调控却少有研究,难以采用基因工程技术来提高嗜热真菌纤维素酶产量。
技术实现要素:
本发明目的在于克服现有技术存在的不足,而提供用于沉默嗜热毁丝霉中腺苷酸环化酶基因的干扰表达载体,本发明采用sirna的dna模板构建热毁丝霉的腺苷酸环化酶干扰表达载体来沉默腺苷酸环化酶,对嗜热毁丝霉纤维素酶基因表达调控进行研究,为后续采用基因工程技术提高嗜热真菌纤维素酶产量提供理论基础。
为实现上述目的,本发明采取的技术方案为:用于沉默嗜热毁丝霉中腺苷酸环化酶基因的sirna的dna模板,其核苷酸序列如seqidno:1~2所示。
另外,本发明还提供实时荧光定量pcr用于检测嗜热毁丝霉中腺苷酸环化酶基因表达量的引物对,其核苷酸序列如seqidno:3~4所示。
另外,本发明还提供一种用于沉默嗜热毁丝霉中腺苷酸环化酶基因的试剂盒,其包括用于沉默嗜热毁丝霉中腺苷酸环化酶基因的sirna的dna模板,其核苷酸序列如seqidno:1~2所示。
作为上述技术方案的改进,试剂盒还包括实时荧光定量pcr用于检测嗜热毁丝霉中腺苷酸环化酶基因表达量的引物对,其核苷酸序列如seqidno:3~8所示。
另外,本发明还提供一种腺苷酸环化酶基因干扰表达载体,其包括sirna的dna模板,其核苷酸序列如seqidno:1~2所示。
作为上述技术方案的改进,腺苷酸环化酶基因干扰表达载体包括mtppdc-ac-mttpdc干扰表达盒,所述mtppdc-ac-mttpdc干扰表达盒的核苷酸序列如seqidno:9所示。
另外,本发明还提供所述的腺苷酸环化酶基因干扰表达载体的构建方法,其包括以下步骤:noti酶和xbai酶切后的puc19-mtppdc-mttpdc质粒和退火后的sirna的dna模板采用t4ligase进行连接。
另外,本发明还提供所述的腺苷酸环化酶基因干扰表达载体在降低滤纸酶、β-葡萄糖苷酶或内切β-1,4-葡聚糖苷酶的活性中的应用。
另外,本发明还提供所述的腺苷酸环化酶基因干扰表达载体在降低滤纸酶、β-葡萄糖苷酶基因blg2或内切β-1,4-葡聚糖酶基因egl1的表达量中的应用。
另外,本发明还提供所述的腺苷酸环化酶基因干扰表达载体在降低磷酸二酯酶基因的表达量中的应用。
本发明的有益效果在于:本发明提供用于沉默嗜热毁丝霉中腺苷酸环化酶基因的干扰表达载体,本发明对嗜热毁丝霉腺苷酸环化酶基因(ac)进行干扰,设计出3种ac基因的干扰片段,双酶切puc19-mtppdc-mttpdc载体和退火后的干扰片段进行连接,获得重组表达载体(即干扰表达载体);将重组表达载体转入嗜热毁丝霉原生质体中,仅阳性菌株ac1的干扰效果良好(ac1中sirna的dna模板是sirna-ac1):ac基因mrna的表达量分下降43%;fpa酶活下降43%,cmc酶活下降了24%,bg酶活下降20%;bgl2、egl1的表达量分别下降24%、53%,pdemrna表达量是野生型wt的33%。
附图说明
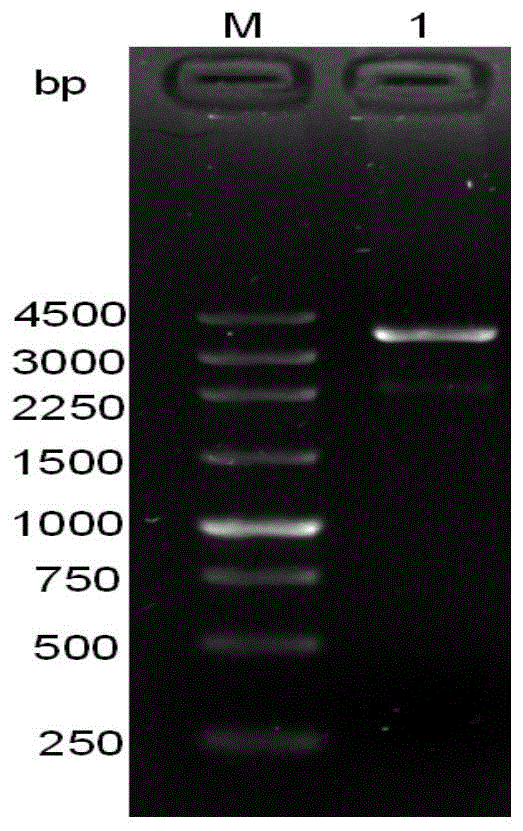
图1为puc19-mtppdc-mttpdc质粒的双酶切电泳图;
图2腺苷酸环化酶基因的干扰表达载体的图谱;
图3显示12个标准转化子的pcr验证结果:干扰表达载体是否成功导入大肠杆菌中;
图4显示转化子的pcr验证结果:pan7-1质粒和干扰表达载体是否成功导入嗜热毁丝霉的原生质体;其中,1:ac1,2:ac2c,3:ac2-1,4:ac2-2,5:ac2-3,6:ac3-1,7:ac3-3,1中sirna的dna模板是sirna-ac1,2~5中sirna的dna模板是sirna-ac2,6~7中sirna的dna模板是sirna-ac3;
图5显示基本培养基第4天,嗜热毁丝霉孢子ac基因mrna表达水量;
图6显示微晶纤维素(1%)诱导条件下野生型和转化子的纤维素酶活,其中纤维酶活包括fpa酶活、cmc酶活和bg酶活;
图7显示野生型和转化子主要纤维素酶基因的表达水平;
图8显示wt和ac1菌株中pde基因的表达水平。
具体实施方式
为更好地说明本发明的目的、技术方案和优点,下面将结合具体实验和附图对本发明作进一步说明。
需要说明的是,本发明存在同一质粒采用多个名字成,如1)重组表达载体等同于干扰表达载体,2)puc19-ac干扰表达载体等同于puc19-mtppdc-mtac-mttpdc干扰表达载体。
实验材料
菌株与质粒
大肠杆菌(e.colijm107):从本学院苟德明教授课题组获得;嗜热毁丝霉(m.thermophilaatcc42464):从美国菌种收藏中心(atcc)处购买;pan7-1质粒:含有筛选真菌的潮霉素抗性基因hphr;筛选大肠杆菌的标记氨苄青霉素抗性基因apr的puc19-mtppdc-mttpdc质粒:由深圳大学构建获得,其以嗜热毁丝霉基因组dna为模板,用引物m-ppdc-f和m-ppdc-r扩增丙酮酸脱羧酶基因(pdc)的启动子mtppdc(pdc基因起始密码子上游587bp序列),用引物m-tpdc-f和m-tpdc-r扩增pdc基因的终止子mttpdc(pdc终止密码子下游357bp序列);接着用hindiii和bamhi对mtppdc进行双酶切,插入经同样酶切的puc19中,转化大肠杆菌jm107;得到的重组质粒puc19-mtppdc经bamhi和ecori双酶切后,与同样酶切的mttpdc连接起来,得到通用型表达载体puc19-mtppdc-mttpdc(参考wangj,wuy,gongy,etal.enhancingxylanaseproductioninthethermophilicfungusmyceliophthorathermophilabyhomologousoverexpressionofmtxyr1[j].journalofindustrialmicrobiology&biotechnology,2015,42(9):1233-1241)。
培养基与试剂
试剂和试剂盒:从takara公司购买的dna限制性内切酶xbai、noti、dnamarker、rna提取试剂(rnaisoplus)、反转录试剂盒(primescriptreagentkit)、荧光定量试剂盒
嗜热毁丝霉原生质体再生培养基:2.5×基本培养基(不含葡萄糖)400ml/l,10×葡萄糖(2g/10ml)100ml/l,2×stc500ml/l,均单独灭菌,使用前分别按4:1:5的比例混合后使用。
stc:218.6g山梨醇,50ml的1mol/lcacl2,10ml的1mol/ltris·cl(ph7.5),混匀溶解后加水定容至1l。
基本培养基(非诱导培养基):mandels营养盐浓缩液100ml,mandels微量元素浓缩液1.0ml,蛋白胨1.0g,无水葡萄糖20.0g,1mol/l的柠檬酸缓冲液50ml,吐温-801.5ml,加水搅拌至完全溶解后,再定容至1l,于121℃高压灭菌25min后保存及备用。
诱导产酶培养基:mandels营养盐浓缩液200ml,mandels微量元素浓缩液1.0ml,葡萄糖3g,蛋白胨1.0g,1mol/l的柠檬酸缓冲液50ml,吐温-801.5ml,以及不同浓度的诱导物,加水搅拌至完全溶解后,再定容至1l,于121℃高压灭菌25min后保存备用。
1%(w/v)纤维二糖诱导培养基:称取0.5g纤维二糖于50ml诱导产酶培养基,加水搅拌至完全溶解后,再定容至1l,于121℃高压灭菌25min后保存备用。
1%(w/v)微晶纤维素诱导培养基:称取0.5g微晶纤维素于50ml诱导产酶培养基,加水搅拌至完全溶解后,再定容至1l,于121℃高压灭菌25min后保存备用。
寡核苷酸引物
实验所用到的寡核苷酸引物见表1和表2。
表1用于验证puc19-ac干扰表达载体完整性的引物
实验方法
腺苷酸环化酶基因ac的sirna的dna模板的设计及合成
从ncbi数据库中获取嗜热毁丝霉的ac基因序列。在设计sirna的dna模板核苷酸的网站(https://rnaidesigner.lifetechnologies.com/orinvitrogenblock-itrnaidesigner),设计筛选出3条干扰序列用于后续实验,见表3。
表3ac基因的sirna的dna模板
注:大斜粗体部分noti和xbai的限制性酶切末点,双下划线部分为茎环结构,其两边是互补的寡核苷酸片段。
sirna寡核苷酸片段退火
将表3中设计好的sirna的dna模板的正义链和反义链送至生工生物工程(上海)公司合成寡核苷酸片段。将其用双蒸水配置成50μm进行退火,其体系如表4。
表4sirna的dna模板的退火反应体系
依照表4中的体系,向标记好的pcr管中加入各试剂并混匀后,在pcr仪中进行退火反应。反应程序如下:95℃变性2min,设置使pcr仪以0.1℃/s下降至25℃(约90min);4℃保存。取出后保存于-20℃,用于后续实验。
质粒提取及酶切
将含puc19-mtppdc-mttpdc的大肠杆菌e.coli在lb平板划线接种,37℃倒置培养12h。然后挑选单菌落接种到适量(30ml)液体lb培养基中,37℃,250rpm培养12h。如果需提取含有氨苄青霉素抗性基因apr的质粒,lb平板和液体培养基均需加入100μg/ml的氨苄青霉素。
用上海生工的质粒提取试剂盒提取质粒,具体操作步骤参照该试剂盒说明书,提取质粒后测定浓度和纯度,选取质量良好的质粒作好标记,-20℃冰箱保存,用于后续实验。
puc19-mtppdc-mttpdc质粒上含有noti/xbai的酶切位点,用快切酶noti和xbai于37℃双酶切质粒1-2小时,用金属浴的方法在80℃失活2分钟,使用上海生工纯化试剂盒进行纯化。
腺苷酸环化酶基因干扰表达载体构建
用noti和xbai酶切后的puc19-mtppdc-mttpdc质粒和退火后的ac基因sirna的dna片段采用t4ligase进行连接,体系参照takarat4ligase说明书。使用下面步骤进行干扰表达载体的构建即标准转化:
(1)-80℃冰箱中取出感受态e.colijm107放置在冰上进行融化;
(2)一般每管感受态为100μl,所以向每管感受态中加入100pg-10ng待转化质粒,缓慢轻柔混匀,在冰上静置30min;
(3)水浴锅预先调至42℃,将离心管放置其中温育90s,之后立即放在冰上静置3min;
(4)向每管中加入800μl预热至37℃的lb液体培养基(无抗生素)颠倒混匀;
(5)37℃,200rpm振荡培养1h;
(6)分别取不同量的菌液(1-2μl、10-20μl、100-200μl)涂布于含apr的lb平板上,37℃倒置培养12-14h;
(7)在含apr的lb平板上,分别挑取单菌落于10μl的无菌水中,做好标记,设计验证引物,分别取其中1μl进行菌落pcr验证,体系如下:
表5菌落pcr验证体系
反应程序为:94℃5min;94℃30s,55℃30s,72℃1min,30个循环;72℃7min;4℃保存;
(8)电泳检测菌落pcr产物后,发现有目的条带的,利用剩余9μl的菌液进行扩大培养(30mllb液体培养基,37℃250rpm12-16h),再用上海生工的试剂盒提取质粒,将质粒送去测序,经过序列比对后验证阳性转化子,结果正确的即为所构建的puc19-mtppdc-mtac-mttpdc干扰表达载体,将这些转化子进行保种,提取的质粒保存于-20℃冰箱,用于后续实验。
嗜热毁丝霉原生质体制备与转化
(1)从-80℃冰箱中取出嗜热毁丝霉保存菌种,2.5μl接在pda平板正中间于45℃培养箱中倒置培养6d;
(2)提前灭好菌的0.2%吐温-80洗涤并制备孢子悬浮液,需要充分分开孢子,避免结团现象的发生,用血球计数板计算所制备孢子悬浮液的浓度;
(3)吸取1×108个孢子加入到30ml左右的液体基本培养基中,在45℃,200rpm/min的条件下震荡培养12h左右,在显微镜下镜检孢子萌发的情况,当看到大部分孢子萌发时,就可以进行下一步实验;
(4)将所有菌液加入到50ml的无菌离心管中,5000r/min离心10min,在无菌工作台中弃去上清;
(5)向离心管中加入10ml的1mol/lmgso4溶液,洗涤沉淀后,5000r/min离心10min,弃去上清,重复一遍;
(6)在重复第(5)步骤的时候,配制10ml的酶解液,配制方法为:称取100mgsigma的溶壁酶(sigma),在工作台中加入10ml的1mol/lmgso4溶液,充分溶解后,采用过滤除菌的方法获得无菌酶解液;
(7)第(6)步配好的10ml酶解液全部加入到离心后的沉淀中,使沉淀悬浮,在28℃,70r/min的培养条件下缓慢震荡培养2h,显微镜下观察其酶解的情况;
(8)加入10mlstc溶液到酶解液中,上下颠倒使其充分混匀,5000r/min离心5min,倒掉上清;
(9)再向沉淀中加入20mlstc溶液洗涤原生质体后5000r/min离心5min,倒掉上清;重复此步骤一次;
(10)加入1ml左右的stc重悬原生质体;
(11)吸取200μl溶液到无菌1.5ml离心管中,向其中加入10μg所构建的puc19-mtppdc-mtac-mttpdc干扰表达载体和5μg的pan7-1质粒,然后轻轻混匀;
(12)将1.5ml离心管放到提前预热到57℃的金属浴中热激2min;
(13)向其中加入50μl的60%peg4000溶液,在室温下静置20min;将1.5ml离心管中的溶液转移到新的无菌50ml离心管中,向其中加入2ml的60%peg4000溶液后,混匀,在室温下静置5min;
(14)向50ml离心管中加入20mlstc溶液,5000rpm离心15min,尽量去掉上清;
(15)加入1ml左右的stc重悬沉淀后,将其加到10ml的原生质体液体再生培养基中;在37℃,70rpm条件下缓慢震荡培养40h;原生质体液体再生培养基成分如下:4ml的2.5×基本本培养基,1ml10×葡萄糖,5ml2×stc;
(16)培养后的溶液中加入20ml的stc后混匀,5000rpm离心10min,去掉上清,尽量将上清去掉干净,可以用枪头吸取剩余上清至废液桶中;
(17)沉淀中加入1mlstc溶液使悬浮,将悬浮液涂布到5个含有40μg/ml潮霉素b的pda平板上,45℃恒温培养箱中培养4d左右,观察是否长出白色菌落;将单个的白色菌落挑选至新的含潮霉素b的pda平板上,45℃恒温培养4d左右;
(18)将第二次筛选平板上长出的白色菌落接种到普通pda平板上,45℃恒温培养6d左右,收集成熟孢子保种,用于后续的实验。
转化子基因组dna提取及验证
嗜热毁丝霉原生质体转化获得的转化子,在45℃培养箱中培养6d后,分别用无菌0.2%吐温-80洗涤孢子,制备孢子悬浮液,接种到30ml液体基本培养基中,培养3-4d。使用上海生工生物工程公司的真菌基因组dna提取试剂盒,提取各个转化子的基因组。所提基因组按照表6的体系进行pcr验证。
表6pcr验证
反应程序为:94℃5min;94℃30s,55℃30s,72℃1min,30个循环;72℃7min;4℃保存;
将pcr产物进行电泳检测,使用250bpmarker,将发现有目的条带的pcr产物送至上海生工生物公司进行测序,测序结果比对确定正确的阳性转化子,进行后续的实验。
嗜热毁丝霉总rna提取
将待提取的嗜热毁丝霉成熟孢子使用无菌0.2%吐温-80制备成孢子悬浮液,接种到30ml液体基本培养基或者诱导产酶培养基中,45℃,200rpm震荡培养2-3d。将全部菌液倒入50ml离心管中进行离心,尽可能的弃去上清液,留下菌丝沉淀。用粉红色rnaisoplus试剂(takara)与传统方法结合来提取总rna。rna提取步骤如下:
(1)将菌丝体置于液氮中充分研磨至白色粉末后,转移2-3勺于无rnase的1.5ml离心管中;
(2)向每管离心管中加入1mlrnaisoplus试剂,震荡使充分混匀后于室温下静置5min;
(3)每管离心管中加入200μl氯仿,上下颠倒混匀15s后于室温下静置2-3min;
(4)使用提前预冷到4℃的离心机,转速为12000rpm离心5min后液体明显变为3层,取上清液600μl左右加到新的离心管中,尽可能不要吸到其它层的液体;
(5)转移好的离心管中加入600μl的异丙醇,上下颠倒混匀后,在室温下放置10min;
(6)4℃,12000rpm离心5-7min后,倒掉上清;
(7)每管加入1ml75%乙醇,漩涡震荡30s或者用枪头吹打至沉淀悬浮;
(8)4℃,9000rpm离心5-7min后,倒掉上清,用枪头尽量将上清液吸干净,然后于室温下静置干燥10-15min;
(9)每管中加入30-50μlrnase-freewater,吸取3μl到200μl离心管中,用来测量rna浓度和琼脂糖凝胶电泳检测rna的完整性。剩余rna长期保存在-80℃冰箱中,以供后续实验。
rt-qpcr
提取rna之后,用反转录试剂盒(primescriptreagentkit)去除其中的基因组dna,然后将去除基因组后的rna反转录为cdna。以反转录后的cdna为模板,采用荧光定量试剂盒
荧光定量检测后,采用2-△△ct的方法来计算样品中相关目的基因的相对表达量。基因的表达水平是以beta-tμbμlin基因(genbank:aeo58945.1)作为内参进行对照。
表7去除基因组dna反应体系
在pcr仪中进行反应,其反应程序为:42℃,2min;4℃,∞。
表8rt-pcr的反应体系
反应程序为:37℃,15min;85℃,5s;4℃,∞。此步反应最后得到的溶液即为cdna,-20℃冰箱进行长期保存。一般,将cdna稀释10倍后进行qpcr。
表9qpcr的反应体系
表10qpcr的反应程序
纤维素酶活性的测定
分别制备嗜热毁丝霉野生型和转化子的孢子悬浮液,用血球计数板计数,分别吸取1×108个孢子接种到50ml的1%微晶纤维素诱导培养基、1%纤维二糖诱导培养基、1%玉米芯粉诱导培养基,45℃,200rpm恒温震荡培养。从第3d开始取发酵上清液至离心管中,13000rpm离心5min后吸取上清液至新的离心管中进行相关酶活的测定。
滤纸酶活(filterpaperactivity,fpa)、内切β-1.4-葡聚糖苷酶活(egactivity)、β-葡萄糖苷酶活(bgactivity)参照国标eveleighde等和qb2583-2003的酶活测定方法加以修改后测定。在50℃和指定ph(酸性纤维素酶ph为4.8)条件下,1ml酶液在1h内水解底物生产出的相当于1mg葡萄糖的还原糖量,即为1个酶活力单位μ/g(或μ/ml)。fpa、bg和cmc酶活的测定实验中,每一组均设置3个重复。
fpa酶活的测定
fpa酶活的测定所需要的相关试剂及条件如下:底物:1.0cm×3.0cmwhatman定性滤纸和600μl的50mm柠檬酸钠缓冲液/每管;酶液:150μl/每管,空白对照先不加入酶液。水解反应条件:50℃,1h。终止反应:900μldns/每管。显色反应:100℃沸水浴中煮沸5min,于冷水(冰水)中降温后进行点样,每管3个孔,每孔200μl。空白对照:加入dns后,煮沸之前向空白对照组中加入相应酶液150μl/每管。多功能酶标仪测定:测定每个小孔a540nm处的吸光值。测定a540nm处的吸光值,以空白管吸光值为对照,根据fpa-dns葡萄糖浓度标准曲线和滤纸酶活的计算公式,计算出其酶活值。
cmc酶活的测定
cmc酶活的测定所需要的相关试剂及条件如下:底物:1%cmc-na溶液,500μl/每管。酶液:150μl/每管,空白对照先不加入酶液。水解反应条件:50℃,30min。终止反应:900μldns/每管。显色反应:100℃沸水浴中煮沸5min,于冷水(冰水)中降温后进行点样,每管3个孔,每孔200μl。空白对照:加入dns后,煮沸之前向空白对照组中加入相应酶液150μl/每管。多功能酶标仪测定:测定每个小孔a540nm处的吸光值。测定a540nm处的吸光值后,以空白管吸光值为对照,根据cmc-dns葡萄糖浓度标准曲线和cmc酶活的计算公式,计算出其酶活值。
bg酶活的测定
bg酶活的测定所需要的相关试剂及条件如下:底物:1%水杨苷溶液,150μl/每管。酶液:150μl/每管,空白对照先不加入酶液。水解反应条件:50℃,30min。终止及显色反应:100℃沸水浴中煮沸5min,于冷水(冰水)中降温后进行点样,每管3个孔,每孔200μl。空白对照:加入dns后,煮沸之前向空白对照组中加入相应酶液150μl/每管。多功能酶标仪测定:测定每个小孔a540nm处的吸光值。酶标板,每个离心管的反应液需设置3个重复孔。测定好a540nm处的吸光值,以空白管吸光值为对照,根据bg-dns葡萄糖浓度标准曲线和bg酶活的计算公式,计算出其酶活值。
实验结果与分析
ac干扰序列的设计及重组载体的构建
从ncbi数据库中获取嗜热毁丝霉的ac基因序列,在网站(https://rnaidesigner.lifetechnologies.com/orinvitrogenblock-itrnaidesigner)设计合成sirna-ac1、sirna-ac2、sirna-ac3,经过退火反应成双链后,分别连接到puc19-mtppdc-mttpdc质粒上。not1和xba1双酶切puc19-mtppdc-mttpdc质粒和构建完整的干扰表达载体如图1和图2所示。
分别将完整的3个干扰表达载体转入感受态大肠杆菌jm107中,利用氨苄青霉素进行筛选得到了12个标准转化子,这些标准转化子中可能含有所需要的重组表达载体。利用引物f-ppdc和r-tpdc进行pcr扩增,来验证干扰片段是否转入到重组载体中,所得12个标准转化子是否为阳性转化子。
筛选得到的标准转化子经过pcr验证的结果如图3所示,标号为1从左至右分别是含sirna-ac1重组表达载体的转化子1-1,1-2,1-3,1-4;标号为2从左至右的分别是含sirna-ac2重组表达载体的转化子2-1,2-2,2-3,2-4;标号为3从左至右的分别是含sirna-ac3重组表达载体的转化子3-1,3-2,3-3,3-4。这12个转化子在1000bp的位置处都显示出了目的条带。将这些转化子进行扩大培养后,提取质粒,送至上海生工公司进行测序检测,测序比对结果表明1-3,1-4,2-3,2-4,3-2,3-3这6个转化为阳性转化子,对其保种并进行后续实验。
嗜热毁丝霉原生质体转化及阳性转化子的筛选鉴定
将培养6d的嗜热毁丝霉孢子制备成孢子悬浮液,使其在适宜条件下进行萌发生长一定时间,制备嗜热毁丝霉的原生质体。将含有潮霉素抗性的pan7-1质粒和构建成功的干扰表达载体(质粒),共同转化到嗜热毁丝霉的原生质体中。
在含有潮霉素抗性的pda平板上经过2-3次筛选后,得到7个单克隆转化子。在液体培养基中,45℃,200rpm培养3d左右,提取基因组。以这些基因组dna为模板,使用f-ppdc和r-tpdc引物进行pcr来验证所构建的干扰表达载体是否转入到这些转化子中。将pcr产物进行电泳检测,使用250bpmarker,如果有目的片段,则应出现在1000bp的位置。
pcr产物电泳检测见图4,明显可以看到ac1、ac2c、ac2-1、ac2-2这4个转化子所显现的目的条带很清晰,而ac2-3、ac3-1和ac3-3这3个转化子虽然在1000bp的位置上也有条带但是条带较弱。将这7个转化子的pcr产物送去上海生工公司进行测序,所得测序结果进过比对后发现ac1,ac2-1,ac2-3和ac2c这4个转化子的序列可以完全正确,说明这4个转化子为阳性转化子,将它们进行保种用于后续实验。
ac基因的荧光定量分析
将从菌株wt、ac1、ac2-1、ac2-3和ac2c提取的rna使用反转录试剂盒进行反转录后,以反转录得到的cdna为模板进行qpcr。以tμb基因为内参基因,利用设计好的ac基因的qpcr引物来检测4个转化子中ac基因mrna表达量,从而检测转化子中ac基因的干扰情况。
使用steponesoftware软件来分析结果,以野生型wt为对照,设定其ac基因mrna的表达量为1,从而检测ac1、ac2-1、ac2-3和ac2c转化子ac基因mrna的表达量。如图5所示,与野生型菌株ac基因mrna的表达量相比,转化子ac1和ac2-3中ac基因mrna的表达量分别下降了43%和34%;选择转化子ac1和ac2-3进行后续的酶活测定等实验。
纤维素酶活的测定
分别取嗜热毁丝霉野生型wt、ac1和ac2-3三个菌株的新鲜孢子制备孢子悬浮液,使浓度为108个,接入含1%微晶纤维素的诱导培养基(50ml),45℃,200rpm,振荡培养。定时取3-7d的菌液进行fpa、bg、cmc酶活的测定。
如图6所示,经过数据分析发现,在培养第4d时,与wt酶活相比,ac1的fpa酶活下降了43%,cmc酶活下降了24%,bg酶活下降了20%;而ac2-3的fpa、cmc、bg酶活与wt各酶活相比没有明显下降。
主要纤维素酶基因的荧光定量分析
使用1%微晶纤维素培养基,培养嗜热毁丝霉野生型wt及转化子ac1、ac2-3三个菌株。fpa、bg、cmc酶活测定结果,发现干扰菌株ac1的酶活效果与野生型wt相比,降低的比较明显,所以以野生型wt为对照组,检测干扰菌株ac1的相关纤维素酶基因mrna的表达情况。
将wt和ac1孢子液,分别接种到pda平板上,45℃倒置恒温培养6d。制备wt和ac1菌株的新鲜孢子悬浮液接种到1%微晶纤维素培养基中,45℃,200rpm,振荡培养4d,提取wt和ac1菌株的rna。
使用反转录试剂盒反转录rna,获得的cdna为模板,检测wt和ac1几种纤维素酶基因以及调控因子mrna表达水平差异。以tμb为内参基因,主要检测bgl1,bgl2,bgl3,egl1,egl3,cre1,ace1,xyr1。使用steponesoftware软件分析,设定wt菌株各个基因mrna表达量为1,计算出ac1菌株各基因的mrna表达量。如图7所示,ac1菌株中bgl2,egl1的表达量与wt相比,分别下降了24%和53%;而其他基因的表达量与wt相比,没有降低甚至有明显增加,如egl3。
磷酸二酯酶基因的荧光定量分析
磷酸二酯酶可以水解camp,它和腺苷酸环化酶共同调节胞内camp水平。分别提取野生型wt和干扰菌株ac1的rna,使用反转录试剂盒反转录rna,获得的cdna为模板,以tμb为内参基因,检测磷酸二酯酶pde基因在两个菌株之间的表达量差异。
设定wt菌株pdemrna表达量为1,计算出ac1菌株pdemrna表达量。如图8所示,在干扰菌株ac1中,pdemrna表达量是野生型wt的33%。
本发明对嗜热毁丝霉腺苷酸环化酶ac基因进行干扰,设计3种ac基因的干扰片段,双酶切puc19-mtppdc-mttpdc载体和退火后的干扰片段进行连接,获得重组表达载体。将重组表达载体转入嗜热毁丝霉原生质体中,仅菌株ac1的干扰效果较好(ac1中sirna是sirna-ac1):ac基因mrna的表达量分下降43%;fpa酶活下降43%,cmc酶活下降了24%,bg酶活下降20%;bgl2、egl1的表达量分别下降24%、53%,pdemrna表达量是野生型wt的33%。
最后所应当说明的是,以上实施例用以说明本发明的技术方案而非对本发明保护范围的限制,尽管参照较佳实施例对本发明作了详细说明,本领域的普通技术人员应当理解,可以对本发明的技术方案进行修改或者同等替换,而不脱离本发明技术方案的实质和范围。
sequencelisting
<110>深圳大学
<120>用于沉默嗜热毁丝霉中腺苷酸环化酶基因的干扰表达载体
<130>2019.04.14
<160>9
<170>patentinversion3.3
<210>1
<211>51
<212>dna
<213>人工合成
<400>1
ggccgcgatgatctcgaggacaattcaagagattgtcctcgagatcatcgc51
<210>2
<211>51
<212>dna
<213>人工合成
<400>2
ctaggcgatgatctcgaggacaatctcttgaattgtcctcgagatcatcgc51
<210>3
<211>21
<212>dna
<213>人工合成
<400>3
gctgccgatggaattgtggtg21
<210>4
<211>22
<212>dna
<213>人工合成
<400>4
ccgaagactgcttgctgttgac22
<210>5
<211>22
<212>dna
<213>人工合成
<400>5
ggcttacggctccaaggacaag22
<210>6
<211>22
<212>dna
<213>人工合成
<400>6
gcgggaagttgaaccaggacat22
<210>7
<211>22
<212>dna
<213>人工合成
<400>7
gatcgcactcgcacagctacag22
<210>8
<211>22
<212>dna
<213>人工合成
<400>8
gcaatgtcatcggcttcctggt22
<210>9
<211>935
<212>dna
<213>人工合成
<400>9
tgcctcttggcgggagccgcccgataactagtataactagttgtaactccgtatccggtt60
acggaaacggaaaggcccgctcggctgttctccggcggctccccgatcgctgatcagagc120
atggaacagatgtcaattacatcactcccgcgtaaacgaaccatagttatcgaaccacag180
ttatcgaaccacagagccagcccatgggaacgtctgaacagctcggaggatgcaaccgat240
attgcaatgcaaaacgtcacccatgctacaattaattccctgcacaactacttgtaagcc300
gcgaggcctagaacacagttgcagaacctgggtatcgtgcctgtggtctgatgcagatat360
gtgtcaccactcaagaccccgccaacacgccgcttcgaggccctgaacagtacaaagggc420
gcttcaaattcgtacaagcccccccgaggccgttttcaagtctttgtatgaccatctatt480
ttccgattgacgtccctcacggattctctttcgttgctgacctccttgtgaccacaaaca540
tcgccaacaacagacgcggccgcaactcgctcaagtacaattcaagagattgtacttgag600
cgagttgcctagagatggtggcttggtggctgaaggaggagaaatggcggagcggacagt660
ccggtgtcggctgcggtgatgagaaatctctttcttaattgagccatagtgaaataaggg720
actgagctgccttgctgtgtttggccattctcaacttcagtgacacatttccctccctcc780
ctaccaccttgtctcgttttctcttgggattgctatgtattttctgaacaaaggggcgca840
ggaaaaggaggtggtaacacgtgactcgttctcggagtccatctgcagcggctcttgtcc900
tcgtttctcaagtgctccacacaaccagccaacac935
- 还没有人留言评论。精彩留言会获得点赞!