一种sgRNA、敲除载体、KLF4基因的敲除方法及其应用与流程
本发明属于基因工程技术领域,具体涉及crispr/cas9的应用技术,也涉及猪小肠上皮细胞系(ipec-j2)基因敲除载体、基因敲除方法及其应用。
背景技术:
crispr/cas9技术是来源于细菌和古生菌中的一种由sgrna(smallguiderna)介导的适应性免疫系统。其中sgrna用于识别、结合靶点dna,当cas9蛋白活性被激活后可结合区域下游的dna并对其进行切割,生成双链dna断裂,诱发由非同源末端连接修复机制造成的移码突变,从而实现对靶基因的编辑。crispr/cas9技术因其简便和高效的特性,近几年来作为真核基因进行定点编辑的重要遗传手段在人类及动植物基因功能研究领域得到广泛应用。
猪小肠上皮细胞(ipec-j2)分离自新生仔猪空肠中段的柱状上皮细胞,具有研究猪病原体感染的物种特异性,是研究猪多种疾病研究的理想体外模型,尤其在产肠毒素大肠杆菌和病毒性腹泻抗性基因功能鉴定研究中广泛应用。
klf4为kruppel样转录因子家族成员,参与调控细胞增殖、分化、胚胎发育等重要生命过程,并介导先天免疫反应。目前尚未有猪klf4基因的敲除载体及klf4基因敲除的小肠上皮细胞模型。
技术实现要素:
发明目的:为了克服现有技术中的不足,本发明所要解决的技术问题是提供了一种sgrna。
本发明还要解决的技术问题是提供了一种双链dna。
本发明还要解决的技术问题是提供了一种载体或细胞。
本发明最后要解决的技术问题是提供了一种klf4基因的敲除方法。
技术方案:为了解决上述技术问题,本发明所采用的技术方案是:一种sgrna,所述sgrna的碱基序列为acagccatgtcagactcgcc。
本发明内容还包括一种双链dna,所述双链dna的上游序列为:
sgrna3f:
双链dna的下游序列为:
sgrna3r:
本发明内容还包含所述的sgrna或所述的双链dna的载体,该载体是一种可打靶猪klf4基因的crispr/cas9敲除载体。
本发明内容还包含所述的载体的细胞。
本发明内容还包含一种klf4基因的敲除方法,包括以下步骤:
1)靶位点设计及sgrna序列的合成:根据猪klf4基因找出cds区5’端进行敲除靶位点设计sgrna向导序列;
2)打靶载体的构建:将设计的sgrna向导序列退火形成双链dna序列,然后与载体相连接得到连接产物,将连接产物导入大肠杆菌得到菌落,菌落提取质粒得到敲除载体,根据敲除载体及sgrna序列设计pcr引物,对菌落pcr的产物进一步测序验证获得阳性打靶载体;
3)细胞转染:将阳性打靶载体加入处于对数生长期的ipec-j2细胞的悬浮液中进行电转得到细胞液;
4)klf4基因的敲除:将嘌呤霉素对细胞液进行药物筛选,将获得的单克隆细胞转移扩大培养,当细胞长满时,分离出部分细胞并提取其基因组dna,根据klf4基因全序列,在敲除靶位点附近设计高特异性的引物,然后对提取的基因组dna进行pcr扩增得到pcr产物,对产物进行验证即可。
其中,所述步骤2)中的pcr引物为:
f3:
r3:
其中,所述步骤2)中菌落pcr的反应程序为95℃10min;95℃35sec,60℃35sec,72℃35sec,35个循环;72℃10min。
其中,所述步骤3)中ipec-j2细胞浓度为1~2×106个/ml。
其中,所述步骤4)高特异性的引物序列为:
f:
r:
其中,所述步骤4)pcr反应程序为95℃5min;95℃10sec,60℃10sec,72℃10sec,35个循环;72℃5min。
本发明的敲除方法具体包括以下步骤:
1)将5’端添加caccg的sgrna序列
2)将双链dna与线性化的敲除载体pgk2.1连接,得到连接产物;
3)以引物:
f1:
r1:
f2:
r2:
f3:
r3:
对连接产物进行菌落pcr筛选,经扩大培养和质粒提取,取得阳性打靶载体;
4)将阳性打靶载体转染ipec-j2细胞,提取细胞基因组dna,在敲除靶位点附近设计高特异性的引物,进行pcr扩增;
5)cruisertm酶切pcr产物筛选获得可高效敲除klf4基因的1条sgrna;
6)pcr产物测序验证,获得klf4基因敲除的ipec-j2细胞。
本发明采用crispr/cas9技术敲除ipec-j2细胞系基因组中klf4基因序列,建立的klf4基因敲除的ipec-j2细胞可为深入揭示klf4基因在疾病抗性中的作用机制及其在猪抗病育种中的应用提供更直接有效的研究模型。
有益效果:本发明相对于现有技术,具有以下优点:
1、首次利用crispr/cas9技术构建klf4敲除的ipec-j2细胞系,可作为猪疾病研究的理想细胞模型;
2、利用crispr/cas9技术进行基因敲除,比基因沉默、干扰等技术手段可更有效地敲除klf4基因,更有助于klf4蛋白的功能研究;
3、基因测序检测表明klf4基因序列被敲除,可造成klf4基因功能的缺失,是较为理想的klf4基因敲除的ipec-j2细胞模型。
4、本发明的敲除方法简便,所提供的sgrna序列可对靶基因实现高效率敲除。
附图说明
图1为菌落pcr凝胶电泳检测图;
图2为菌落pcr产物测序峰图:图2a为sgrna1对应打靶载体测序峰图;图2b为sgrna2对应打靶载体测序峰图;图2c为sgrna3对应打靶载体测序峰图;
图3为敲除靶位点pcr扩增凝胶电泳检测图;
图4为cruisertm酶切筛选klf4基因敲除细胞dnapcr产物凝胶电泳检测图;
图5为cruisertm酶切筛选klf4基因敲除细胞dnapcr产物测序峰图;
图6为klf4基因敲除细胞dna序列与klf4基因未敲除细胞dna序列对比分析结果。
具体实施方式
下面结合附图对本发明作更进一步的说明。
实施例1
1.靶位点设计及sgrna序列的合成:
根据ncbi数据库获取猪klf4基因(geneid:595111)的转录本cds序列,并找出cds区5’端进行敲除靶位点设计,应用crisprdesign(http://crispr.mit.edu/)设计了三条sgrna向导序列:
在三条sgrna向导序列sgrna1:
三对sgrna序列如下:
sgrna1f:
sgrna1r:
sgrna2f:
sgrna2r:
sgrna3f:
sgrna3r:
2.打靶载体的构建:
(1)将以上正、负链sgrna序列进行退火形成双链dna序列,反应体系如下:
将上述反应体系瞬时离心并置于95℃孵育3min,自然冷却20min。
(2)利用bbsi酶对敲除载体pgk2.1进行酶切线性化,反应体系如下:
37℃孵育4h,试剂盒纯化回收线性化的载体pgk2.1。
(3)取上述退火形成的双链dna序列与线性化的载体pgk2.1进行连接,反应体系如下:
将上述体系瞬时离心后,16℃孵育30min,获得连接产物。
(4)将10μl以上连接产物加入dh5α感受态,轻轻混匀,置于冰上孵育30min;42℃热激60sec,迅速拿出置冰上冷却2~3min;向管中加入800μl无抗soc液体培养基,摇床(37℃/160rpm)培养45min;4500rpm离心5min,弃去600μl上清,将沉淀重悬于剩余的200μl上清中,均匀涂布于含卡那霉素抗性的培养平板上,倒置37℃过夜培养。
根据敲除载体pgk2.1及三条sgrna序列设计三对pcr引物:
f1:
r1:
f2:
r2:
f3:
r3:
菌落pcr的筛选:从以上培养板中挑取白色单克隆菌落并置于pcr管中,向pcr管中添加上述上、下游引物各1μl,缓冲液(10x)2μl,dntp2μl,taq酶0.2μl,双蒸水13.8μl。pcr反应程序为95℃10min;95℃35sec,60℃35sec,72℃35sec,35个循环;72℃10min。
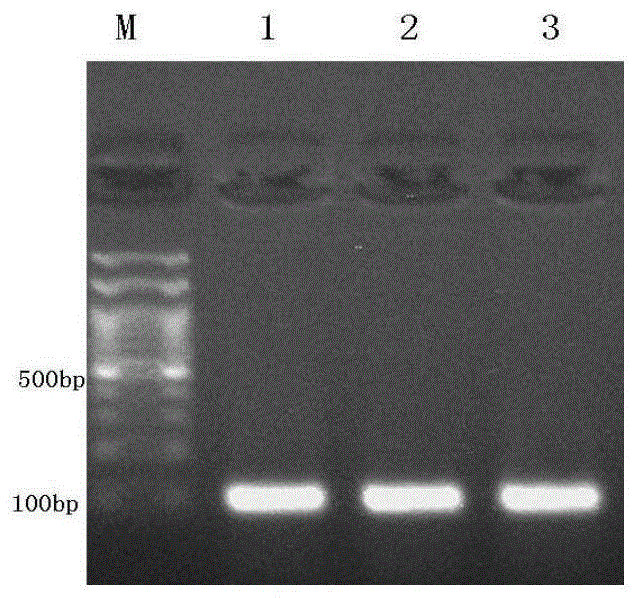
图1为菌落pcr凝胶电泳检测图,其中,m为100bpdnamarker,1、2、3为阳性菌落pcr条带。由图1可见:以上方法获得了阳性打靶载体。
对得到的阳性菌落pcr产物进一步测序验证,如图2所示,阳性打靶载体基因序列与pgk2.1载体及sgrna序列相吻合。对阳性打靶载体采用常规方法进行扩大培养,进行质粒提取,用于后续实验。
3.细胞转染:
将处于对数生长期的约2×106个猪小肠上皮细胞(ipec-j2)置于15ml离心管,1000rpm/5min离心弃上清,用210μldpbs悬浮细胞,转移至1.5ml离心管中。将5~8μg上述3种打靶载体分别加入上述含有ipec-j2细胞悬浮液的1.5ml离心管中,轻轻混匀,将混合液用专用电转枪头转移至电击杯中,待液面凸起后,盖上电击杯盖并至于电转仪,650v/30ms进行电转,之后将细胞液转移至六孔板培养基中。
4.klf4基因的敲除:
电转72小时后,以3μg/ml浓度的嘌呤霉素进行药筛,并用有限稀释法稀释细胞至10块96孔板中,在37℃的co2培养箱中静置培养7天后,观察单克隆生长情况,约14天后将单克隆细胞转移至48孔中扩大培养。当细胞长满48孔时,分离出部分细胞,使用dna提取试剂盒提取其基因组dna。
5.pcr扩增步骤4中提取的dna:
根据klf4基因全序列,在敲除靶位点附近设计高特异性的引物,序列为:
f:
r:
pcr扩增反应体系如下:
pcr反应程序为95℃5min;95℃10sec,60℃10sec,72℃10sec,35个循环;72℃5min,95℃3min,获得长度为326bp的pcr产物。
图3为敲除靶位点pcr扩增凝胶电泳检测图。其中,m为100bpdnamarker,1、2、3为pcr产物条带。由图3可见:敲除靶位点附近设计的引物可对目的序列进行成功扩增。
6.klf4基因敲除细胞的筛选与验证:
(1)cruisertm酶切筛选klf4基因敲除细胞
使用识别错配的cruisertm酶对上述长度为326bp的pcr产物进行酶切检测,配置反应体系如下:
45℃反应20min,向上述10μl反应体系中加入2μl6×stopbuffer,之后进行琼脂糖凝胶电泳检测。图4为cruisertm酶切筛选klf4基因敲除细胞结果,其中m代表100bpdnamarker;1-1、1-2代表转染sgrna1打靶载体的细胞;2-1、2-2代表转染sgrna2打靶载体的细胞;3-1、3-2代表转染sgrna3打靶载体的细胞。由图4可知,sgrna1和sgrna2不能对klf4基因进行有效敲除,而sgrna3可有效敲除klf4基因。根据灰度值进行打靶效率分析,结果显示,3-1和3-2所对应细胞中klf4基因打靶效率分别为54.8%、56.1%。
(2)klf4基因敲除细胞dnapcr产物测序验证:
对cruisertm酶切筛选出来的klf4基因敲除细胞dnapcr产物进行测序验证,如图5测序峰图所示,确定转染sgrna3打靶载体的细胞为klf4基因敲除细胞。对klf4基因敲除细胞dna做ta克隆后送测序,与klf4基因未敲除细胞dna序列进行比对,发生敲除的细胞dna序列突变为116bp和137bp的缺失(图6)。
图6为ta克隆测序分析klf4基因敲除序列。其中,第一行为klf4基因未发生敲除的序列,第二三行为klf4基因敲除序列。
以上所述仅是本发明的优选实施方式,应当指出:对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
序列表
<110>扬州大学
<120>一种sgrna、敲除载体、klf4基因的敲除方法及其应用
<160>17
<170>siposequencelisting1.0
<210>1
<211>20
<212>dna
<213>sgrna1(artificialsequence)
<400>1
acgttattcggggcacctgc20
<210>2
<211>19
<212>dna
<213>sgrna2(artificialsequence)
<400>2
acaacactcacgttattcg19
<210>3
<211>20
<212>dna
<213>sgrna3(artificialsequence)
<400>3
acagccatgtcagactcgcc20
<210>4
<211>24
<212>dna
<213>f1(artificialsequence)
<400>4
catatgcttaccgtaacttgaaag24
<210>5
<211>21
<212>dna
<213>r1(artificialsequence)
<400>5
gcaggtgccccgaataacgtc21
<210>6
<211>24
<212>dna
<213>f2(artificialsequence)
<400>6
catatgcttaccgtaacttgaaag24
<210>7
<211>19
<212>dna
<213>r2(artificialsequence)
<400>7
cgaataacgtgagtgttgt19
<210>8
<211>24
<212>dna
<213>f3(artificialsequence)
<400>8
catatgcttaccgtaacttgaaag24
<210>9
<211>20
<212>dna
<213>r3(artificialsequence)
<400>9
ggcgagtctgacatggctgt20
<210>10
<211>25
<212>dna
<213>sgrna1f(artificialsequence)
<400>10
caccgacgttattcggggcacctgc25
<210>11
<211>25
<212>dna
<213>sgrna1r(artificialsequence)
<400>11
aaacgcaggtgccccgaataacgtc25
<210>12
<211>24
<212>dna
<213>sgrna2f(artificialsequence)
<400>12
caccgacaacactcacgttattcg24
<210>13
<211>23
<212>dna
<213>sgrna2r(artificialsequence)
<400>13
aaaccgaataacgtgagtgttgt23
<210>14
<211>25
<212>dna
<213>sgrna3f(artificialsequence)
<400>14
caccgacagccatgtcagactcgcc25
<210>15
<211>24
<212>dna
<213>sgrna3r(artificialsequence)
<400>15
aaacggcgagtctgacatggctgt24
<210>16
<211>20
<212>dna
<213>f(artificialsequence)
<400>16
ggacttcggaggacctctga20
<210>17
<211>20
<212>dna
<213>r(artificialsequence)
<400>17
ccacccacagaagcccacta20
- 还没有人留言评论。精彩留言会获得点赞!