一种共表达草鱼呼肠孤病毒外衣壳蛋白VP4和VP35的重组杆状病毒的制备方法和应用与流程
本发明属于生物
技术领域:
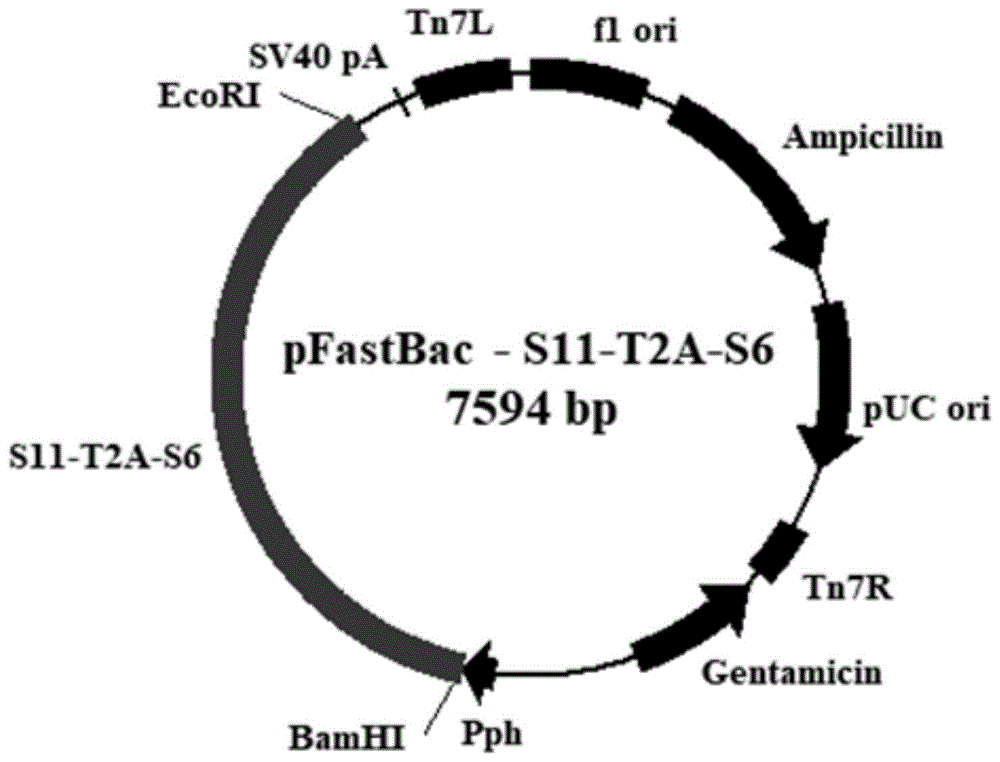
,具体涉及一种共表达草鱼呼肠孤病毒外衣壳蛋白vp4和vp35的重组杆状病毒的制备方法和应用。技术背景草鱼出血病是由草鱼呼肠孤病毒引起的严重危害草鱼养殖的一种病毒性传染病,主要引起患病草鱼各器官、组织不同程度出血,内脏组织坏死,死亡率极高,对我国水产养殖带来巨大危害和经济损失。当前国内主要针对基因ⅰ型草鱼呼肠孤病毒开展研究,而对于基因ⅱ型草鱼呼肠孤病毒的研究较少。根据全国主要草鱼养殖区流行病学调查结果表明,ⅱ型草鱼呼肠孤病毒是我国目前的主要流行株。草鱼呼肠孤病毒隶属于呼肠孤病毒科,水生呼肠孤病毒属,病毒粒子呈二十面体对称的球形体,成熟病毒直径为70-80nm,具有双层衣壳,无囊膜,基因组由11条分节段的双链rna组成,共编码7种结构蛋白和5种非结构蛋白。基于生物信息学分析和前人研究结果,ⅱ型草鱼呼肠孤病毒s6节段编码的vp4蛋白和s11节段编码的vp35蛋白为该病毒的外衣壳蛋白,外衣壳蛋白在病毒进入细胞的过程中起着重要作用,并且能诱导宿主产生免疫应答,具有重要的免疫原性。因此,建立能够高效表达ⅱ型草鱼呼肠孤病毒外衣壳蛋白的表达系统对于草鱼呼肠孤病毒疫苗的研发及草鱼出血病的有效防治具有重要意义。多基因表达载体的优势在于可同时表达2个或多个基因,但早期常用的多基因表达载体多存在容量小,易出现上、下游基因表达失衡,蛋白活性低或所表达蛋白不能精确定位等缺陷。近年,一种名为自剪切2a肽的多基因载体构建策略受到广泛关注。2a肽是首先在小核糖核酸病毒中发现的一种具有“自剪切”功能的肽链。报道称,2a肽剪切反应发生在翻译阶段的核糖体肽基转移酶中心,其剪切功能不是通过蛋白酶水解作用,而是经核糖体“跳跃”来实现的,发生在c末端gly-pro残基之间。研究认为2a肽改变了核糖体的活性,促进2a肽残基gly与trnagly之间酯链的水解,从转录复合物上释放上游多肽的同时又推进下游蛋白的翻译。此过程被认为是核糖体从一个密码子跳跃至下一个密码子而中间没有肽键形成。其功能主要是能够将上下游2个蛋白实现单独翻译。2a肽介导的多基因表达载体具有高裂解活性且上、下游基因等摩尔表达等优点,其剪切效率约为90%,具备明显的优势,是目前较为理想的多基因表达策略,已广泛应用于动物转基因研究。目前大量研究表明该剪切功能只在真核表达系统中发挥,原核表达系统内无此剪切功能。不同的2a肽剪切效率不一致,有可能存在剪切不完全的现象。2a肽上游序列会影响剪切效率。草鱼呼肠孤病毒作为中国分离的第一种鱼类病毒,尚无利用该种自剪切肽在同一真核表达载体中同时表达两个抗原性蛋白的报道。目前有64种密码子,其中61个密码子编码20种氨基酸,3个密码子使蛋白质合成终止,因此,每一种氨基酸至少含有一个密码子(met和trp),最多的含有6个不同的密码子编码同一种氨基酸(arg,leu,和ser)。但是,宿主细胞并不是同等的使用每一个密码子来编码氨基酸的,宿主细胞倾向于用同义密码子的一种或几种,不同的宿主细胞中,密码子的使用具有很大的差异性,即使同一宿主中高表达量的蛋白和低表量的蛋白之间,甚至是同一操纵子下的不同蛋白其密码子的使用也具有很大差异。已经有研究表明,高频使用的密码子虽然能增加核糖体的延伸速度,但是过度表达会导致氨基酸“饥饿,从而改变负载trna的丰度。由于密码子的改变会影响蛋白质的共翻译过程,优化的密码子虽然可以加速核糖体在mrna上的移动,但是可能会影响蛋白质的活性并导致构象改变的蛋白质被水解。因此,在进行密码子优化使其在相应宿主中表达异源蛋白时,需要综合考虑密码子的重复、起始密码子环境、终止密码子环境、酶切位点设计、核糖体结合位点、稀有密码子、gc含量、mrna的稳定性、mrna的二级结构等因素,通过合理有效的密码子优化,以明显提高蛋白表达量。针对上述问题,本发明首次提供了一种共表达草鱼呼肠孤病毒外衣壳蛋白vp4和vp35的重组杆状病毒的制备方法,利用该方法制备的重组病毒,外源表达量高,适用于疫苗的生产。技术实现要素:本发明的目的在于提供了一种共表达草鱼呼肠孤病毒外衣壳蛋白vp4和vp35的重组杆状病毒的制备方法,方法简单。本发明的另一个目的在于提供了上述方法制备的重组杆状病毒在制备草鱼呼肠孤病毒病疫苗中的应用。为了达到上述目的,本发明采取以下技术措施:一种共表达草鱼呼肠孤病毒外衣壳蛋白vp4和vp35的重组杆状病毒的制备方法,是将seqidno.1、seqidno.3和seqidno.2所示序列顺序插入到pfastbactmⅰ的bamhⅰ和ecori酶切位点之间,得到重组质粒pfastbac-s11-t2a-s6;再将pfastbac-s11-t2a-s6的外源基因同源重组到杆状病毒骨架质粒bacmid中,得到bacmid-s11-t2a-s6;利用杆状病毒表达系统,转染sf9细胞获得。上述方法制备的重组杆状病毒在制备草鱼呼肠孤病毒病疫苗中的应用,利用上述方法制备重组杆状病毒,进行规模化扩大培养,可大量获得具有重要免疫原性的草鱼呼肠孤病毒vp4和vp35蛋白,以此作为草鱼呼肠孤病毒亚单位疫苗。与现有技术相比,本发明具有以下优点:1、本发明首次构建了共表达草鱼呼肠孤病毒vp4和vp35蛋白杆状病毒表达系统,可应用于制备亚单位疫苗,具有较高的生产实践意义。2、ⅱ型草鱼呼肠孤病毒s6节段编码的vp4蛋白和s11节段编码的vp35蛋白为该病毒的外衣壳蛋白,外衣壳蛋白在病毒进入细胞的过程中起着重要作用,并且能诱导宿主产生免疫应答,具有重要的免疫原性。利用优化后的中间连接蛋白构建了vp4和vp35共表达载体,实现了vp4和vp35在同一载体中的高效表达,可用于制备新型亚单位疫苗,提高疫苗的免疫保护率。附图说明图1为重组质粒pfastbac-s11-t2a-s6示意图。图2为外衣壳蛋白vp35和vp4的pcr扩增;其中,m:2000bp;泳道1、2:vp4;泳道3、4:vp35。图3为pfastbac-s11-t2a-s6质粒的双酶切(bamhⅰ、ecorⅰ)鉴定示意图;m:5000bp;泳道1:pfastbac-s11-t2a-s6bamhⅰ、ecorⅰ双酶切。图4为蓝白斑筛选重组bacmid-s11-t2a-s6示意图。图5为重组bacmid-s11-t2a-s6的pcr鉴定示意图;其中:m:5000bp;泳道1、2、3、4、5:重组bacmid-s11-t2a-s6;泳道6:阴性对照。图6为正常sf9细胞与重组杆状病毒感染sf9细胞对比示意图;其中左图为正常sf9细胞,右图为重组杆状病毒感染sf9细胞72h。图7为pcr检测杆状病毒中的目的基因示意图;其中:7.1m:1000bp;泳道1、2:vp4;泳道3:阴性对照7.2m:2000bp;泳道1、2:vp35;泳道3:阴性对照。图8为westernblot分析vp4和vp35蛋白在杆状病毒中的表达示意图;其中,m:蛋白预染marker;泳道1、2、3、4、5分别为感染重组杆状病毒p3、p4、p5、p6、p7表达的外衣壳蛋白;泳道6:感染野生型杆状病毒。图9为ifa检测vp4和vp35蛋白在重组杆状病毒中的表达示意图;其中,a:感染重组杆状病毒sf9细胞;b:感染野生型杆状病毒sf9细胞。具体实施方式实施例1:一种共表达草鱼呼肠孤病毒外衣壳蛋白vp4和vp35的重组杆状病毒的制备方法,包括下述步骤:1)外衣壳蛋白vp35和vp4的pcr扩增与t-a克隆载体构建用trizol法提取感染草鱼呼肠孤病毒106株的草鱼卵巢细胞系(gco)总rna,用反转录试剂盒反转录为cdna并以此为模板分别对s6(seqidno.2所示,编码vp4)、s11(seqidno.1所示,编码vp35)基因进行pcr扩增,pcr产物用1%的琼脂糖凝胶电泳检测,结果显示,如图2所示,可分别得到1827bp的vp4和945bp的vp35。用pcr产物纯化回收试剂盒分别对目的片段进行回收纯化。用于扩增vp35的引物序列是:上游引物vp35-f:5’-gggatccatggaaccagcaaaaccattgacg-3’;下游引物vp35-r:5’-agctagcgtgatggtggtgatgatgcatgccggagccggactgtccctggatctcaggtttgaag-3’,上下游酶切位点分别为bamhⅰ和nhei,c端下游加入了ser-gly-ser-gly序列。用于扩增vp4的引物序列是:上游引物vp4-f:5’-agctagcccgaatggactcgcatggatgaaag-3’;下游引物vp4-r:5’-ggaattctctagtgatggtggtgatgatgagacggaggaggccagtatcgagttaatttgt-3’,上下游酶切位点分别为nhei和ecorⅰ。每个蛋白后面均添加了6个his标签。通过重叠pcr扩增的方法,用一个序列长度为75bp、编码25个蛋白的短肽t2a作为linker将二者连接起来。中间连接蛋白t2a序列:atgcattctcgaggctcgggcgaaggacgaggctctttgttaacttgtggcgatgtggaagaaaatcccggccct。然后将此构建好的两个目的基因片段克隆到pgem-t载体的bamhⅰ和ecori位点之间,获得pgem-s11-t2a-s6克隆质粒。蛋白连接顺序为:vp35(310aa)+6xhis+t2a(25aa)+vp4(608aa)+6xhis。2、pfastbac-s11-t2a-s6重组杆状病毒载体的构建通过双酶切,将目的基因从pgem-s11-t2a-s6质粒上切下,并连接到供体质粒pfastbactmⅰ载体中,获得vp35和vp4的杆状病毒表达质粒pfastbac-s11-t2a-s6。经bamhⅰ、ecor酶切和测序鉴定,如图3所示,双酶切可得到两条预期大小约4800bp和2800bp的特异性条带,进一步经测序证实为目的基因序列,表明质粒构建成功,获得了目的基因的阳性重组质粒pfastbac-s11-t2a-s6(图1)。3、重组质粒bacmid-s11-t2a-s6的构建和鉴定参照bac-to-bac杆状病毒表达系统说明书,将构建正确的1ng的pfastbac-s11-t2a-s6转化至含有杆状病毒骨架质粒bacmid的dh10bac感受态细胞中。混匀后置于冰上30min。42℃热激45s,立即冰上2min。加入900μl室温的soc培养基于37℃,225转震荡培养4小时。用soc培养基按10-1、10-2、10-3稀释感受态细胞后,分别取100μl稀释复苏菌液涂布于含有50μg/ml卡那霉素、7μg/ml庆大霉素、10μg/ml四环素、100μg/mlbiuo-gal、40μg/mliptg的lb平板上,37℃倒置培养48h,在helper质粒的辅助作用下,重组转移载体pfastbac-s11-t2a-s6与杆状病毒骨架质粒bacmid发生同源重组,将外源的目的基因整合到杆状病毒骨架质粒中形成重组bacmid质粒,可见有蓝白斑菌落出现,如图4所示。各挑取10个白色阳性单克隆菌落,重新划线在新鲜的含有50μg/ml卡那霉素、7μg/ml庆大霉素、10μg/ml四环素、100μg/mlbiuo-gal、40μg/mliptg的lb平板上,37℃过夜培养。经过三轮蓝白斑筛选纯化阳性菌落。3.1、重组质粒bacmid-s11-t2a-s6的提取(1)使用灭菌的牙签挑白色单克隆进入含有50μg/ml卡那霉素、7μg/ml庆大霉素、10μg/ml四环素的2mllb培养基中,于37℃摇床,250~300rpm振荡培养24h以上。(2)取1.5ml培养物于1.5ml离心管中,14000×g离心1min。(3)吸尽上清液,加入0.3ml的溶液ⅰ(50mmol/l葡萄糖,10mmol/ledta,25mmol/ltris-hcl(ph8.0))用枪头轻轻吹打重悬细胞。(4)加入0.3ml的溶液ⅱ(0.2mol/lnaoh,1%sds,现用现配)轻轻混匀,室温静置5min,溶液变清。(5)缓慢加入0.3ml的溶液ⅲ(3mol/l醋酸钾,ph5.5)混匀,形成蛋白及dna沉淀,冰上放置5~10min。(6)14000g离心10min。(7)轻轻将上清转移至含有0.8ml异丙醇的离心管中,不要吸到白色沉淀。颠倒混匀,冰上放置5~10min。(8)室温14000g离心15min。(9)小心弃去上清,不要浮起沉淀,加入0.5ml70%的乙醇,颠倒离心管洗涤沉淀数次,室温14000g离心5min。(10)尽可能多的弃去上清,不要搅动沉淀。室温5~10min风干沉淀,不要过分干燥。(11)加入40μlte溶液重新溶解dna沉淀。使用pcr分析重组质粒bacmid-s11-t2a-s6由于重组杆粒dna较大,约为135kb,所以对于验证外源基因是否插入不能用传统的限制性内切酶剪切的方法进行分析,杆粒包含puc/m13正负引物位点,可用puc/m13正向引物和puc/m13反向引物对重组bacmid进行pcr鉴定。puc/m13-f:5’-cccagtcacgacgttgtaaaacg-3’puc/m13-r:5’-agcggataacaatttcacacagg-3’pcr反应体系如下:phusiondnapolymerase0.5μl5×phusionhfbuffer10μl10μmolforwardprimer2.5μl10μmolreverseprimer2.5μl10μmoldntps1μl稀释100倍的重组质粒bacmid2μldmso1.5μlnuclease-freewater30μl总体积50μl反应条件如下:98℃30s;98℃10s,55℃30s,72℃2min30s,共30个循环;最后72℃延伸10min。全部反应结束后,取出8μlpcr产物电泳检测,用1%的琼脂糖凝胶电泳检测pcr扩增结果。如图5所示,重组杆粒bacmid-s11-t2a-s6扩增出的目的条带分子量大小约为5000bp,表明成功构建重组杆粒bacmid-s11-t2a-s6。4、重组杆状病毒的获得(1)按照cellfectiontmⅱ脂质体转染试剂盒说明操作,将生长良好的sf9细胞接至6孔板中,细胞密度约8×105个/孔。(2)27℃恒温培养箱静置1h,使细胞贴壁。(3)在这期间制备bacmid-s11-t2a-s6与cellfectiontmⅱreagent复合物。a.在两个1.5ml的ep管中分别加入100μl无双抗无血清sf-900tm-ⅲsfm(1×)备用,并分别标记bacmid和转染试剂。b.在bacmid的ep管中加入2μg组质粒bacmid-s11-t2a-s6,轻弹管壁混匀。在转染试剂ep管中加入6μlcellfectiontmⅱreagent(在使用前将cellfectiontmⅱreagent倒置5~10次使其充分混匀),轻弹管壁混匀,室温静置30min。c.将稀释后的bacmid-s11-t2a-s6和cellfectiontmⅱreagent混在一起(总体积约210μl),轻弹管壁混匀,室温孵育15-30min。d.孵育后将bacmid-s11-t2a-s6和cellfectiontmⅱreagent混合物加入铺好的6孔板中,置于27℃恒温培养箱培养4-6h。(4)去掉含有bacmid-s11-t2a-s6和cellfectiontmⅱreagent混合物混合液的细胞培养基,加入2ml新鲜的sf-900tm-ⅲsfm(1×)培养基,在27℃恒温培养箱培养。每天在倒置显微镜下观察并记录细胞生长情况。细胞的病变将出现三个时期,早期(24h),细胞直径增加,可以看到直径增大了25-50%,细胞核增大,可能充满整个细胞;晚期(24-72h),细胞停止生长,与单个细胞相比,细胞不在增大,出现颗粒体,即病毒芽孢信号,部分细胞从细胞培养瓶上脱离;极晚期(﹥72h),细胞破裂,出现细胞凋亡,细胞从细胞培养瓶上脱离。(5)72h后可看到细胞明显病变,如图6所示,收集病变的细胞及培养液,1000r/min离心5min,收集上清,4℃避光保存,即为p1代重组杆状病毒。5、病毒的扩增因细胞毒株p1代的病毒滴度很低,不可用于后面的相关实验,需将p1代毒再次感染sf9细胞,以获得病毒滴度较高的重组杆状病毒,扩增过程如下:将sf9细胞(约为9×105个)传代至t25细胞瓶培养过夜,p1代病毒按5%体积比感染处于对数生长期的细胞,27℃恒温培养箱培养3-5d,细胞出现明显病变时,用移液枪吹打细胞使其脱落,分装于15ml离心管中,1000r/min离心5min,收集上清,4℃避光保存,即得到p2代病毒,相同方法收取p3、p4、p5代病毒,4℃避光保存,或-80℃长期保存。6、重组昆虫杆状病毒tcid50测定将生长状态良好的sf9细胞铺于96孔细胞培养板中,当细胞密度生长至70%时,取p5代重组杆状病毒,按以下比例进行梯度稀释:101、102、103、104、105、106、107、108、109、1010,弃去细胞培养孔中的培养液,加入100μl各个梯度的病毒稀释液,每个梯度重复8个孔,做好空白对照,每天观察细胞病变情况并做好统计。感染96h后根据细胞孔病变数量采用reed-muench法(lgtcid50=高于50%的稀释对数+距离比例×稀释系数的对数)计算病毒滴度。lgtcid50=-7;tcid50=10-7/0.1ml(p5代病毒)7、dna水平检测重组杆状病毒取细胞病毒p3代的培养上清,抽提病毒dna。提取病毒dna的方法:(参考viraldnakitomega公司)(1)在1.5mlep管中加入250μl病毒悬液,同时加入4μllinearacrylamide、250μlbufferbl、10μlob蛋白酶,涡旋振荡ep管,使试剂充分混匀。(2)将上述ep管放入65℃水浴锅中孵育10min,孵育期间拿出ep管,摇晃混匀样品2次。(3)将260μl无水乙醇加到上述ep管中,并充分混匀。(4)将ep管中的混合物转移至吸附柱中(外面套有2ml收集管),室温8000r/min离心1min。(5)将吸附柱从收集管中取出,弃掉收集管中的液体,再将吸附柱重新装回到收集管后,添加500μlbufferhb到吸附柱,室温8000r/min离心1min。(6)将吸附柱从收集管中取出,弃掉收集管中的液体,再将吸附柱重新装回到收集管后,添加700μldnawashbuffer到吸附柱,室温8000r/min离心1min。(7)将吸附柱从收集管中取出,弃掉收集管中的液体,再将吸附柱重新装回到收集管后,添加500μldnawashbuffer到吸附柱,室温8000r/min离心1min。(8)将吸附柱从收集管中取出,更换新的收集管后,室温15000r/min离心2min。(9)将吸附柱取出后,室温静置10min。(10)将吸附柱放置于新的1.5mlep管,加50μl预热的ddh2o于吸附柱中,室温放置3min。(11)室温8000r/min离心1min。收集ep管中液体,放置于-20℃保存备用。以上述提取的病毒dna为模板,按骤1所用扩增vp4和vp35的上下游引物分别进行pcr,最后用1%琼脂糖凝胶电泳鉴定。结果如图7所示,分别扩增出1827bp和933bp两条带,大小与vp4和vp35基因大小一致,说明vp4和vp35成功重组于杆状病毒中。8、重组蛋白的westernblot分析(1)样品的处理:将生长良好的sf9细胞接至6孔板中,细胞密度约8×105个/孔,当sf9细胞汇合度达到70%时,向每个孔中以接种量moi=1接入p3、p4、p5、p6、p7代的重组杆状病毒,另外设立不接毒的正常sf9细胞做对照。72h后接毒的细胞明显病变,将每个孔中的细胞分别收集起来,弃去培养基上清,细胞沉淀用pbs洗两次,加入100μlripa细胞裂解液(含1%pmsf蛋白酶抑制剂),吹打混匀置于冰上裂解10min。将裂解得到的细胞7000r/min离心5min,收集上清,按1:5的比例加入25μl的5×loadingbuffer,沸水煮样10min,使蛋白质变性,之后7000r/min离心5min,离心后直接上样进行sds-page。(2)上样与电泳:12%的sds-page电泳凝胶,每孔上样20μl,蛋白mark10μl,80v,2h。(3)转膜:先把pvdf膜在甲醇中浸泡活化,在将pvdf膜从下到上依次厚滤纸、pvdf膜、胶、厚滤纸的顺序放置,每一层都赶走气泡,盖上转膜仪盖子,调整电压至20v,30min。(4)封闭:将pvdf膜取下,用tbst清洗两遍,每次5min,放入配好的含5%脱脂奶粉的tbst封闭液中,室温封闭1h。(5)一抗孵育:封闭结束后,在摇床上用tbst洗膜3次,每次10min。把pvdf膜放入已经用pbs按1:1000稀释过的小鼠抗his标签单克隆抗体(anti-6×histagantibodyabcam)溶液中,4℃孵育过夜。(6)二抗孵育:一抗孵育结束后,在摇床上用tbst洗膜3次,每次10min。把pvdf膜放入已经用pbs按1:5000稀释过的二抗(山羊抗小鼠igg-hrp)溶液中,室温孵育1h。(7)化学发光:二抗孵育结束后,在摇床上用tbst洗膜3次,每次10min。再把pvdf膜放入暗盒中,加入ecl发光液在暗室曝光,结果如图8所示,可见两条特异性条带,大小分别为66kd和34kd。9、间接免疫荧光(ifa)鉴定外源基因表达(1)将爬片放入12孔板,将生长状态良好的细胞传至12孔板,铺板3h后细胞汇合度达到70%。(2)移去细胞培养液,向每个孔中以接种量moi=1接入重组杆状病毒,与sf9细胞共同孵育1h后,弃去病毒液,加入1ml新鲜培养基,27℃培养3d,细胞开始病变。(3)摇床上用pbs洗3次,每次5min。(4)固定:室温下每个孔中加500μl4%多聚甲醛固定20min。(5)摇床上用pbst洗3次,每次5min。(6)透化:每孔加入500μl0.2%tritonx-100,室温静置10min。(7)摇床上用pbst洗3次,每次5min。(8)封闭:每孔加入500μl5%bsa,室温封闭1h。(9)摇床上用pbst洗3次,每次5min。(10)一抗孵育:每孔加入500μl小鼠抗his标签单克隆抗体(1:500anti-6×histagantibodyabcam),4℃避光孵育过夜。(11)摇床上用pbst洗3次,每次5min。(12)二抗孵育:每孔加入500μl山羊抗小鼠单克隆抗体(1:1000goatanti-mouseigg(h+l)highlythermofisher),室温避光孵育1h。(13)摇床上避光用pbst洗5次,每次5min。(14)细胞染核:每孔加入300μldapi(4’,6-二脒基-2-苯基吲哚)稀释液(1:300),室温避光染色15min。(15)摇床上避光用pbst洗3次,每次5min。(16)封片:取5μl抗荧光淬灭剂滴于载玻片上,加爬片倒扣于载玻片上,用指甲油涂抹于爬片四周进行封片。(17)通过荧光显微镜观察,结果如图9所示,可看到大量绿色荧光,显示该重组病毒能高效感染sf9细胞并产生外衣壳蛋白。实施例2:重组杆状病毒高表达vp4和vp5蛋白:将sf9细胞(约为9×105个)传代至t25细胞瓶培养,当sf9细胞汇合度达到70%时,向每个孔中以接种量moi=1接入p3代的实施例1制备的重组杆状病毒,提取细胞总蛋白,经亲和层析法纯化后,测定重组目的蛋白的浓度为1.6μg/ml,纯度为90.5%。在本申请中,通过重组蛋白的大量表达纯化发现400~600ml的培养基培养的sf9细胞可纯化出225~338μg的蛋白;而前人(expression,purificationandstructuralanalysisoffunctionalgabatransporter1usingthebaculovirusexpressionsystem)未经优化的杆状病毒表达系统每400~600ml的培养基培养的sf9细胞可纯化出蛋白的量为200~300μg,与其相比,本发明提供的重组杆状病毒对蛋白表达量显著提高。实施例3:剪切肽的不同优化后的密码子的剪切效果:按照实施例1的方式,构建了剪切肽的不同优化后的密码子的重组杆状病毒,并考察蛋白的表达量,具体结果如下表:剪切肽的不同优化后的密码子及蛋白表达结果注:经实验验证,优化t2a(4)序列为最优序列,本专利采用该序列采用亲和层析纯化重组蛋白,进行鱼体免疫保护实验。于实验前周,选取健壮无病的同一批次草鱼鱼种体重25-30g,于水温28℃-30℃中充氧饲育。实验分3组,注射剂量(蛋白量/鱼的体重)为:3μg/g以及空白组、阴性对照组,每组30尾。用pbs稀释融合蛋白至所需浓度、与等体积的弗氏不完全佐剂完全混匀,腹腔注射0.1ml/尾。免疫2周后,根据预实验结果采用半致死浓度1.0×106tcid50ml-1进行攻毒(gcrv106),每尾注射0.1ml。观察2周,每日纪录发病与死亡鱼的数量,统计死亡率。计算免疫保护率免疫保护力(rps)=[(对照组死亡率-免疫组死亡率/对照组死亡率]%。结果表显示,攻毒后,对照组75%发病死亡,死亡鱼肌肉、鲍盖、肠道有轻度充血和出血,少部分鱼体腹腔出血,肝胰脏发白,表现出明显的出血病症状;而免疫组存活率95%,表明用纯化的重组蛋白vp35-vp4免疫草鱼对出血病具有较好的免疫保护作用。序列表<110>中国水产科学研究院长江水产研究所<120>一种共表达草鱼呼肠孤病毒外衣壳蛋白vp4和vp35的重组杆状病毒的制备方法和应用<160>3<170>siposequencelisting1.0<210>1<211>942<212>dna<213>人工序列(artificialsequence)<400>1atggaaccagcaaaaccattgacgtttctggatctcacccgactcaatgaggcaatctgc60tctcgggctctcttctttgataatgataataactgctggtctgtgtctccaatcgcccca120aggcaagccaaattccatgactcagttgtctgtattcgatgtggagcgcctattgacaaa180gttcatgcaatgtcaattccaccaccccctgtgcatggctgcatcccaatgctggatcat240agccaatgggaagatttgtacgagttggctgaggatatgggccgctgtatttggtgggct300aaaaagcaactggtcatctggatggagggaatagtgaatctgaaggctggtaaggtgtat360aatgataatgtgagtaaccgcagtgaatggccagatgaggtgtgggatgaaacatgcaaa420atcttctgtaaatgggcaacgcaaaatcgtgtggcgagccgttggattcagtcaccatca480cgtgtgtacaagtttctttgtgaccaggaaagtaagatgaacatcgatgctctggagcta540tccaaccatcagatttttcaggcaccaccgcaatggcctgagatagctatggctgtcccg600cactggtccccgtccgtgcatgagatgctaaatggtcacaaaatggtgatgattgtgccc660cgcttgtccatgccagtcatatttgatcccgccaacggttacgtcgccccaatctacacc720gcggccatgatcagtctcccatcccaatggtgggtgtcacaatatgtaaaagttcatgga780agccacatggtgccacgtctgtatggtgatgacgtcccaaccttacgttcccgcctgcgg840aatgcttctaccacaacctcccactctcatacgcaactcctgatgctgcctgaagctcaa900tcgaccttcaaacctgagatccagggacaggccggagccgga942<210>2<211>1824<212>dna<213>人工序列(artificialsequence)<400>2ccgaatggactcgcatggatgaaagtcgcaggatcaggtatgggaccaggttcactgcaa60attgtacacgcaacagacgggtcaccgtattgttacatccctccggacgccatgtctagt120atggaaaaatcagcaggtgccgttaatttgtgggaacctctattcgccgctatattcgaa180acctgctctacagtgaacgtgtccgactttactgatgagttcagtgcctacgttggcgtc240agtgaacccgaggccctcaagaagtacatcgagaaaggagtcttcatgtcaacatcacaa300gcgaaaaatttctttgggacactcgggcagaggatggcgaggataaagggttggtctgaa360gatatacggacggcggctgctatgataccagtagacacacctcatggctcggtcacgtgc420gattggaagagcttacttagattttgggacgaaaatctaccagtggataatgtatgcaaa480caataccaaaatgctgtatatgaggtagcattgaagatataccctgcgttacgggccgga540aatagtggatcacttctctctgatgtcgttggcgtattagctggtggtgcggcaggaact600acagcgtctggagtgagtatggtcgaaggtgtgacacgtaatgagcaagtgtctcgaatc660ctgacatcagcagacgttgatattttgacgtcgtcagcgccagtgcctgtctcagctttg720agaactccaccagtggaattgtcctatcaacccagcgccctgagtgccaagaccacccca780tggttggttcgatatcccgggactactgccatagagaagacattcgacgtcgggacaaca840tccaagacgacgtattacctgagcatgggtaactccggtggtggagatttgatgattgat900ctgaagcgactcccagcatgcggacttgaatactccctgcgtggtatcccgatcatatac960gataccaatctgaccgcggcgaagctggctaaggttactcctgcattgctgatgctgcag1020acggctaaaccacttagtgcggagatcaccgccgcagatattcaagctatcacacctctt1080gtagtagggactgataagctcaacactttggtcaccactggttttggcaatattcgcaac1140attaccgacttctcaatgtctgcaatttgggaaccagagacagtcagcgcggcaggcaat1200tactatctatggccgactgtaatcggtgatgcatcaatgacatcagactgggggacaatt1260agcacatctctagctaatggcaggctccgcgtcgcacctctggacctgactaatgcactc1320cacaaaggcaatgtcgtggagtcaatcgtaccatctgatgtgctaggtaatgcctcacca1380gaggaaatgcgttccgcgttaccagcagatgtgctgacagctttcaaagccaagctcaca1440acagtggcttctgtagttggccgtgccttaaaccccaacgacagtgcgcatgcaccatcc1500tccggcaccgtcctcggcccgcttgcaattgaaaacaaggcccaatcgaaacctaaaccc1560gtatcagatctgtggatagctgctcggcgtggtgtgaatctattcgtagcctctccaatc1620gcgagtctgcaaacaggcgtgccggtcatgggggacagcagcgtgttgacaagcttgatg1680ggtggtgcaactaccgcgttgaatactggcgatatgggtaccccaagcctggaggccaca1740gcgaaacgggcagctagagttgctggaggactgctacgacagagagtcatggacaaatta1800actcgatactggcctcctccgtct1824<210>3<211>75<212>dna<213>人工序列(artificialsequence)<400>3atgcattctcgaggctcgggcgaaggacgaggctctttgttaacttgtggcgatgtggaa60gaaaatcccggccct75当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1
- 调控家蚕质型多角体病毒编码的NS5基因的miRNA及其应用
- 检测水稻黑条矮缩病毒的qRT-PCR及其应用
- 一种用于呼肠孤灭活疫苗的佐剂及其制备方法
- 一种猪流行性腹泻病毒的多重rt-pcr检测试剂盒的制作方法
- 一种控制草鱼出血病的中草药饲料添加剂及制备方法
- 抗蓝舌病病毒ns3蛋白的单克隆抗体btv-ns3-3d8及其识别的b细胞表位和应用
- 利用小麦绿矮病植株为毒源建立的一种玉米粗缩病品种抗性接种鉴定方法
- 抗蓝舌病病毒ns1蛋白的群特异性单克隆抗体btv-ns1-1f11及其识别的b细胞表位和应用
- 一种西江病毒SYBR GreenⅠ荧光定量PCR检测方法
- 抗蓝舌病病毒12型ns1蛋白的单克隆抗体btv12-ns1-1f8及其识别的b细胞表位和应用