一种替格瑞洛药用II晶型的制备方法与流程
本发明涉及替格瑞洛的制备方法,具体涉及一种替格瑞洛药用ii晶型的制备方法,具体而言,本发明涉及通过结晶条件的选择,高纯度的制备替格瑞洛不同晶型,该工艺稳定,收率相对较高,工艺中没用到特殊设备与试剂。
发明背景
替格瑞洛(ticagrelor,化学结构见式ⅰ)属于环戊基三唑并嘧啶类化合物,是astrazeneca公司研发的一种新型具有选择性地治疗急性冠状动脉综合征(acs)的小分子抗凝血药物,能可逆性地作用于血管平滑肌细胞上的嘌呤2受体(p2)亚型p2y12,无需代谢激活,对二磷酸腺苷引起的血小板聚集有明显的抑制作用,具有起效迅速,疗效强,显著降低患者出血事件风险,降低心血管病死率等特点。
阿斯利康公司的专利cn1247583c公开了替格瑞洛的四个晶型(晶型i,晶型ii,晶型iii,晶型iv)和无定形状态及其制备方法。在上述晶型中,ⅱ型结晶稳定性最佳,同时也为原研单位上市药品所具有晶型,但ⅱ型结晶的结晶溶剂氯仿毒性较大,不适合规模化生产;且结晶颗粒不均匀,存在大颗粒(40-60um),在进行制剂制备时,需先进行粉碎,制剂成本高。
综上,仍有必要对替格瑞洛结晶工艺进行进一步研究,找到晶型纯度高、粒径小且均匀,同时可工业化生产、生产成本更低的替格瑞洛结晶工艺。
技术实现要素:
发明目的
本发明的目的在于提供一种制备替格瑞洛不同晶型的新方法,该方法工艺稳定、晶型纯度高、粒径小且均匀,易于规模化生产。
技术方案
本发明提供了一种晶型纯度高、粒径小且均匀、适易于产业化生产的的制备方法,主要包括以下步骤:
一种通过2-(3ar,4s,6r,6as)-6-(5-胺基-6-氯-2-丙硫基-4-嘧啶基)氨基四氢-2,2-二甲基-4h-环戊烯并-1,3-二氧杂环戊烷-4-基]氧]-乙醇制备替格瑞洛的新方法,该方法的特征是:
步骤(1):2-(3ar,4s,6r,6as)-6-(5-胺基-6-氯-2-丙硫基-4-嘧啶基)氨基四氢-2,2-二甲基
-4h-环戊烯并-1,3-二氧杂环戊烷-4-基]氧]-乙醇在亚硝酸钠、乙酸存在条件下,在一定的
温度下反应生成2-[[(3ar,4s,6r,6as)-6-[7-氯-5-丙硫基-3h-1,2,3-三唑[4,5-d]嘧啶-3-基]四
氢-2,2-二甲基-4h-环戊烯并-1,3-二氧杂环戊烷-4-基]氧]-乙醇(中间体2);
步骤(2):中间体2在盐酸甲醇溶液作用下生成替格瑞洛粗品;
步骤(3):替格瑞洛粗品浓缩后,加入惰性溶剂,生成替格瑞洛成品。
所述的步骤(3)中加入惰性溶剂时残余物温度40~70℃,其中优选为60~70℃。
所述的步骤(3)中浓缩后残余物体积/中间体2=0-20v/w,优选6-8v/w。
步骤(3)中惰性溶剂为正庚烷、环己烷和异辛烷,其中优选正庚烷。
所述的步骤(3)的析晶温度-10~10℃,其中优选为0~10℃。
其制备路线为:
本发明的有益效果是,通过简单的工艺制备,高收率的获得粒径小而均匀的替格瑞洛,且无氯仿适合于规模化生产。因为析晶条不同,即不同溶剂或者析晶溶剂比例对得到的晶型有影响,本发明中实施例中显示有的是晶型i,有的是晶型ii,有的是晶型i和ii混晶,细微条件改变其晶型也随之发生改变,具体来说结晶处理过程中浓缩的时间和浓缩程度,滴加不同溶剂等,其结果不同,故本发明提供一种最佳优化的条件获得纯度高、收率高、其粒径小(低于40μm)和定向获得晶型ii的替格瑞洛产品。
附图说明
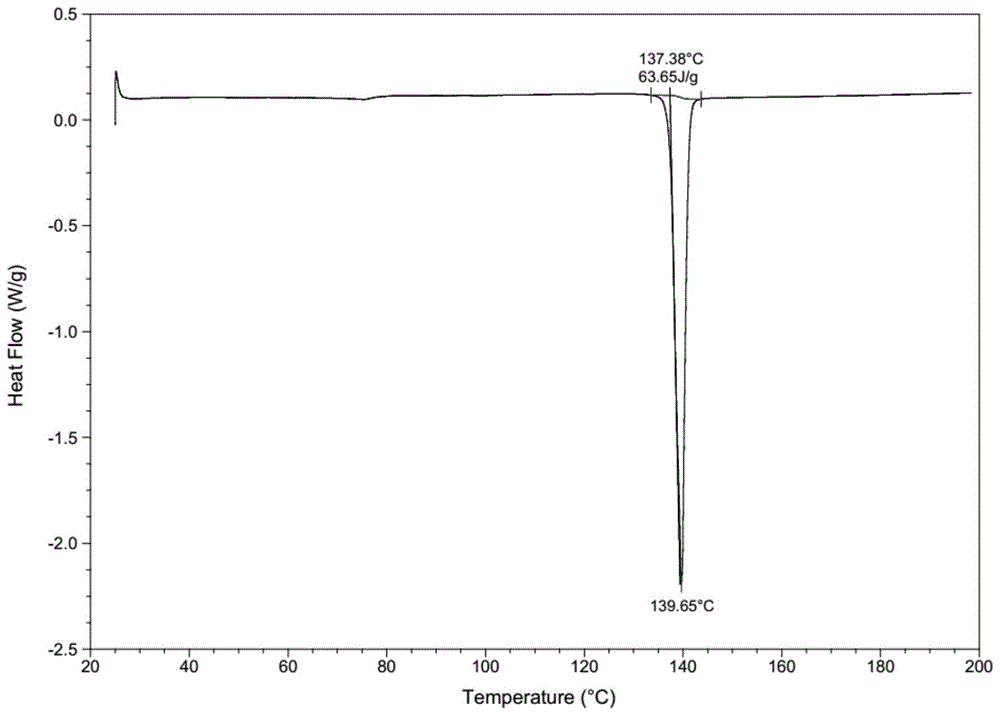
图1替格瑞洛ii晶dsc图
图2替格瑞洛i晶dsc图
图3替格瑞洛i和ii混晶dsc图
图4替格瑞洛ii晶xrd图
图5替格瑞洛i和ii混晶xrd图
具体实施方式
为了更好地理解本发明,下面结合具体事例来详细说明本发明的技术方案,但是本发明并不局限于此。
实施例1
于1l反应瓶中,依次加入甲苯(300ml)、2-(3ar,4s,6r,6as)-6-(5-胺基-6-氯-2-丙硫基-4-嘧啶基)氨基四氢-2,2-二甲基-4h-环戊烯并-1,3-二氧杂环戊烷-4-基]氧]-乙醇(中间体1)(62.6g,149mmol)、乙酸(50.0g,834mmol),搅拌降温至0-10℃,滴加亚硝酸钠水溶液(10.8g亚硝酸钠/25ml水)。滴毕后升温至20-30℃,搅拌10min,滴加碳酸钾水溶液(61.8g碳酸钾/125ml水),调节ph=7-8,体系静置分液,有机相备用(中间体2)。
于3l反应瓶中,加入上步有机相,搅拌降温至0-10℃,滴加预冷的浓盐酸和甲醇的混合液(271.9g浓盐酸/250ml甲醇),搅拌反应5min,静置分液。向水相中加入1l纯化水和0.5l乙酸乙酯,降温至0-10℃,向反应体系中加入125g碳酸氢钠,调ph=7-8,静置分液。水相再用0.5l乙酸乙酯萃取,合并有机相,再用1.2l的纯化水洗涤两次,静置分液,有机相用活性炭脱色和无水硫酸钠干燥30min,过滤备用(替格瑞洛有机相);
有机相于2l反应瓶中40-45℃浓缩至剩余约0.5l,残余物升温至60-70℃,控温滴加正庚烷,滴毕后,降至0-10℃,搅拌3h,离心,滤饼40-45℃真空干燥至恒重(差重小于0.5%),得到类白色固体,收率范围:80-90%。
经hplc检测,产品纯度为99.5%;dsc和xrd检测,样品为ii晶型;马尔文粒度仪检测粒度范围小于30um。
实施例2
替格瑞洛有机相溶液制备方法同实施例1。
有机相于2l反应瓶中滴加正庚烷,滴毕后,降至0-10℃,搅拌48h,离心,滤饼40-45℃真空干燥至恒重(差重小于0.5%),得到类白色固体,收率范围:60-65%。
经hplc检测,产品纯度为99.2%;dsc和xrd检测,样品为ii晶型;马尔文粒度仪检测粒度范围小于20um。
实施例3
替格瑞洛有机相溶液制备方法同实施例1。
有机相于2l反应瓶中40-45℃浓缩至剩余约0.5l,残余物升温至60-70℃,控温滴加环己烷,滴毕后,降至0-10℃,搅拌3h,离心,滤饼40-45℃真空干燥至恒重(差重小于0.5%),得到类白色固体,收率范围:75-80%。
经hplc检测,产品纯度为99.5%;dsc和xrd检测,样品为ii晶型;马尔文粒度仪检测粒度范围30-40um。
实施例4
替格瑞洛有机相溶液制备方法同实施例1。
有机相于2l反应瓶中40-45℃浓缩至剩余约0.5l,残余物升温至60-70℃,控温滴加异辛烷,滴毕后,降至0-10℃,搅拌3h,离心,滤饼40-45℃真空干燥至恒重(差重小于0.5%),得到类白色固体,收率范围:70-80%。
经hplc检测,产品纯度为99.3%;dsc和xrd检测,样品为ii晶型;马尔文粒度仪检测粒度范围30-40um。
实施例5
替格瑞洛有机相溶液制备方法同实施例1。
有机相于2l反应瓶中40-45℃浓缩至剩余约0.5l,残余物升温至60-70℃,控温滴加正庚烷,滴毕后,降至-10-0℃,搅拌2h,离心,滤饼40-45℃真空干燥至恒重(差重小于0.5%),得到类白色固体,收率范围:80-90%。
经hplc检测,产品纯度为95.6%;dsc和xrd检测,样品为ii晶型;马尔文粒度仪检测粒度范围30-40um。
对照例1
替格瑞洛有机相溶液制备方法同实施例1。
有机相于2l反应瓶中40-45℃浓缩至干,残余物升温至60-70℃,控温滴加正庚烷,滴毕后,降至0-10℃,搅拌3h,离心,滤饼40-45℃真空干燥至恒重(差重小于0.5%),得到类白色固体,收率范围:80-90%。
经hplc检测,产品纯度为99.5%;dsc和xrd检测,样品为i和ii混晶;马尔文粒度仪检测粒度范围40-50um。
对照例2替格瑞洛有机相溶液制备方法同实施例1。
有机相于2l反应瓶中40-45℃浓缩至剩余约0.5l,控温滴加正庚烷,滴毕后,降至至0-10℃,搅拌3h,离心,滤饼40-45℃真空干燥至恒重(差重小于0.5%),得到类白色固体,收率范围:80-90%。
经hplc检测,产品纯度为97.2%;dsc和xrd检测,样品为i和ii混晶;马尔文粒度仪检测粒度范围40-50um。
对照例3
按照原研专利cn1247583c中实施例2重复。
将氯仿(150ml)加入到45g替格瑞洛中,加热至溶解,将所得溶液放置结晶过夜,并在流动氮气下40-50℃干燥至恒重(差重小于0.5%),得到类白色固体,收率范围:75-85%。经hplc检测,产品纯度为98.2%;dsc和xrd检测,样品ii晶体;马尔文粒度仪检测粒度范围40-60um,且有大颗粒。
- 还没有人留言评论。精彩留言会获得点赞!