一种NGS建库分子接头及其制备方法和用途与流程
本发明属于分子生物学领域,涉及一种ngs建库分子接头及其制备方法和用途,尤其涉及一种ngs建库分子条形码接头及其制备方法和用途,具体涉及一种单分子标签扩增子建库接头及其制备方法和用途。
背景技术:
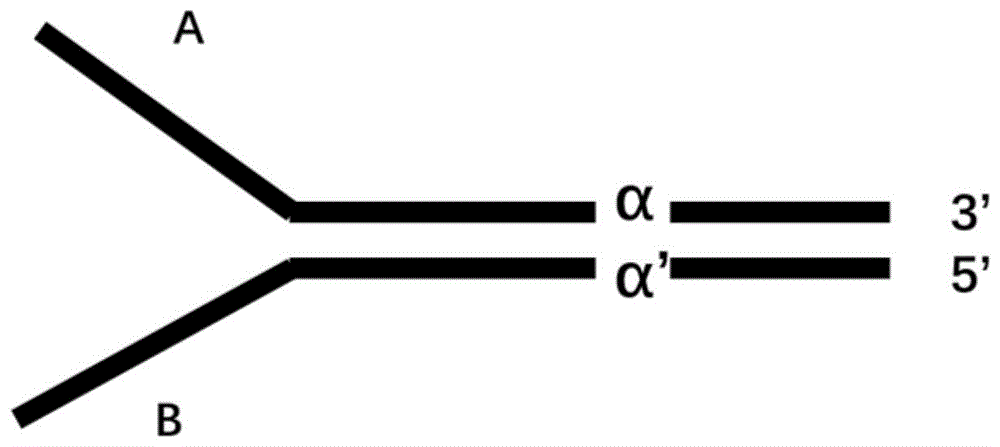
:二代测序技术已经成为成熟有效的分子检测手段,应用大规模平行测序的策略,具备同时检测多个靶标和多个样品的能力,二代测序技术具有一代测序和pcr方法不可比拟的通量优势。采用高深度测序的方法,二代测序在肿瘤检测、产前诊断和病原体筛查方面都有广泛的应用。通过极高深度测序,二代测序可以达到极高的检测灵敏度。使得微小残留病灶检测,肿瘤突变液体活检、肿瘤早筛,以及可诉肿瘤突变负荷在治疗过程中的监控评估成为可能。然而极高深度测序亦存在其局限性,由于建库需要多轮的pcr扩增过程,底物分子片段在扩增过程中,由于扩增酶导致的的复制错误,会引入大量非自然的突变,在加上测序过程中的信号错误,会严重提升高深度测序的假阳性率。建库扩增加上测序过程中的团簇生长以及测序过程中的光信号错误,大约有1%的错误率,而很多检测领域如ctdna突变一般会在1%以下。需要采取相应的手段降低假阳性,否则会显著降低检测结果的可靠程度。目前主要从建库方法设计和数据过滤方面来消除以上所述因素造成的影响。有代表性的是在建库接头中引入单分子识别标签。如采用多个随机兼并碱基构成的单分子标记标签。例如通过引物引入14个兼并碱基的单链分子标签可以使得错误率降低20倍。而在扩增前基于连接反应引入单分子标签的方法则更有利于去除pcr扩增偏好性带来的影响,有利于检测底物拷贝数变化和rna测序的表达丰度。基于连接反应引入单分子标签的代表性方法为illumina的y字形单分子标签接头,该技术同时将两个独立的长兼并序列单分子标签连接到底物双分子的两端。通过一对通用引物进行扩增后分别得到两组来自底物分子的序列。这种依赖通用引物复制底物分子的原理并不适用于基于特异性基因引物延伸的富集建库。另外,由于y型duplex测序底物双分子两端都连接了长随机碱基标签,存在由于测序过程中固定的错误率导致的留存率过低的缺点,要达到足够符合分析要求的reads需要额外大幅度的提高测序深度,增加了使得测序成本。现有单分子标签接头由两部构成,如图1所示,a、b代表两个引物结合区;α和α'代表两个互相匹配的兼并序列互补对区,通过引物结合区和兼并序列区的组合作为一个特有的单分子标签。接头通过连接反应结合到底物分子上后形成如图2所示的结构;y字形接头的制备工艺如图3所示,需要经过双链退火延伸及酶切后纯化的步骤。现有的y字形接头的制备过程繁琐,基本原理是两个长短不同的单链序列通过共有的退火匹配序列进行退火然后进行延伸实现随机分子标签的复制,然后进行酶切和纯化。由于存在随机序列的自由匹配干扰,使得两个单链序列正确退火的效率非常低,这就要求非常严格的变性胶纯化程序,从而增加了接头制备的难度和延长了制备时间。现有的duplex建库接头及其实现工艺的局限性限制了其在实际科研或者医学诊断中的应用。因此,提供一种适用于扩增子建库的单分子标记测序接头和相匹配的制备工艺具有重要意义和广阔的应用前景。技术实现要素:针对现有技术的不足及实际的需求,本发明提供一种ngs建库分子接头及其制备方法和用途,本发明的分子接头适用于扩增子建库,将相同的分子标签所有reads都包含的突变位点判定为真正的生物学突变,显著降低假阳性概率。为达此目的,本发明采用以下技术方案:第一方面,本发明提供一种ngs建库分子接头,所述分子接头包括5’端引物结合区、单分子标签区和共有连接区,所述共有连接区包括第一序列和第二序列,所述第一序列和第二序列互补连接。本发明的接头分子连接底物后,在建库过程中沿着5’到3’端的方向延伸,补齐末端突出的部分,基于特异性基因引物延伸的富集建库,可用于扩增子建库,适用范围广。优选地,所述5’端引物结合区的碱基数为10-40个,例如可以是10个、11个、12个、13个、14个、15个、16个、17个、18个、19个、20个、21个、22个、23个、24个、25个、26个、27个、28个、29个、30个、31个、32个、33个、34个、35个、36个、37个、38个、39个或40个,优选为25-35个。优选地,所述单分子标签区由随机碱基构成。本发明中,通过加入随机碱基构成的单分子标签,有效区分来自不同模板的底物,具有相同标签的所有reads都包含的突变位点才判定为真正的生物学突变,显著降低假阳性概率。通过单分子标签可以有效的区别rna表达量、基因组拷贝数扩增/缺失与测序深度不均衡造成的测序区域reads数量差异,提升目的片段量性检测的可靠度。优选地,所述随机碱基的个数为6-14个,例如可以是6个、7个、8个、9个、10个、11个、12个、13个或14个,优选为8-12个。优选地,所述共有连接区的互补碱基对数为4-8对,例如可以是4对、5对、6对、7对或8对。本发明中,共有连接区的互补碱基对数为4-8对,若共有连接区过短则影响接头双链的稳定性,若过长则影响后续上机测序的质量评估,也会降低有效reads长度。优选地,所述第一序列的3’末端突出一个粘性末端胸腺嘧啶。本发明中,分子接头的第一序列3’末端突出一个粘性末端胸腺嘧啶,与待测底物分子上的腺嘌呤互补结合,完成分子接头与底物的连接。优选地,所述胸腺嘧啶经过羟基化处理。优选地,所述第二序列的5’端进行磷酸化处理。优选地,所述5’端引物结合区的核苷酸序列如seqidno.2所示,具体为5’-gtctcgtgggctcggagatgtgctcttccgatct-3’;所述单分子标签区为12个随机碱基构成;所述共有连接区的第一序列如seqidno.3所示,具体为5’-tgactgtagaaga-3’;所述共有连接区的第二序列如seqidno.4所示,具体为5’-tcttctacagtca-3’。第二方面,本发明提供一种用于制备如第一方面所述的分子接头的单链多核苷酸,所述单链多核苷酸按顺序依次包括引物结合区、单分子标签区、第一互补序列区、环形连接区和第二互补序列区。优选地,所述单分子标签区由随机碱基构成。优选地,所述第一互补序列区与第二互补序列区的碱基互补。优选地,所述第一互补序列区和第二互补序列区包含相同的酶切位点。优选地,所述环形连接区的碱基个数为20-60个,例如可以是20个、21个、23个、25个、28个、30个、32个、33个、34个、35个、36个、37个、38个、39个、40个、41个、42个、43个、44个、45个、46个、47个、48个、49个、50个、52个、53个、55个、56个、58个、59个或60个,优选为30-40个。优选地,所述环形连接区的核苷酸上具备生物素标记。优选地,所述生物素标记的核苷酸个数不少于一个,例如可以是1个、2个、3个、4个、5个、6个、7个、8个、9个、10个、11个、12个或13个等,优选为1-5个,进一步优选为2个。优选地,所述单链多核苷酸的核苷酸序列如seqidno.1所示,具体如下:gtctcgtgggctcggagatgtgctcttccgatctnnnnnnnnnnnntgactgtagaaga-gcttgcagtgggcu*u*acatggcgatagctagact-tcttctacagtca;其中第1-34bp为5’端引物结合区,35-46bp为单分子标签区,47-59bp为第一互补序列区,60-93bp为环形连接区,94-106bp为第二互补序列区,*表示生物素标记,此处为对两个u进行生物素修饰。第三方面,本发明提供一种制备如第一方面所述的分子接头的方法,所述方法包括如下步骤:(1)将第二方面所述的单链多核苷酸退火;(2)加入限制性内切酶,酶切反应;(3)利用磁珠负选,纯化回收步骤(2)酶切产物。现有的y字形接头通常是两个长短不同的单链序列通过共有退火匹配序列进行退火,然后进行延伸,实现随机分子标签的复制,然后进行酶切纯化,由于存在随机序列的自由匹配干扰,使得两个单链序列正确退火的效率非常低,需要非常严格的变性胶纯化程序,因此整个制备工艺复杂且难度高。本发明的分子接头通过单链多核苷酸退火形成发卡式结构,同时有少量由两条原料分子退火形成的链式结构,经过限制性酶切后,接头原料分子在酶切位点处断裂,形成不带生物素标记的成品接头和带有生物素标记的部分;另外,未发生酶切反应或者部分酶切的原料分子也带有生物素标记,利用带有链霉素的磁珠对反应体系内的核酸分子进行负选,酶切下的核酸条带和未发生酶切反应的原料分子都会被去除,留下高纯度的产物接头结构序列,经过定量、沉淀步骤后即得到成品接头产物。优选地,步骤(1)所述退火的步骤为:a)设置pcr仪程序,94-96℃,4-6min;b)在步骤a)结束后,设置每分钟降温1-2℃,降温时间40-70min。优选地,步骤a)之前还包括预处理步骤:设置热盖温度为100-110℃,例如可以是100℃、101℃、102℃、103℃、104℃、105℃、106℃、107℃、108℃、109℃或110℃,优选为105℃。优选地,所述退火的步骤具体包括以下步骤:设置pcr仪热盖105℃,设置程序95℃,5min;设置每分钟降温1℃至60min。优选地,步骤(3)之后还包括后处理步骤:将纯化回收的酶切产物进行5’端磷酸化处理。优选地,步骤(2)所述限制性内切酶包括hpych4iii、hpy188i或ital中的任一种或至少两种的组合,优选为hpych4iii。优选地,步骤(2)所述酶切的时间为10-14h,例如可以是10h、11h、12h、13h或14h,优选为12h。优选地,步骤(2)所述限制性内切酶的终浓度为0.4-0.8u/μl,例如可以是0.4u/μl、0.5u/μl、0.6u/μl、0.7u/μl或0.8u/μl,优选为0.5u/μl。第四方面,本发明提供一种如第一方面所述的分子接头用于ngs建库和/或测序中的用途。优选地,所述分子接头用于ngs测序的方法包括如下步骤:(1’)将待测样品dna片段化,修复处理,纯化回收;(2’)将第一方面所述的分子接头与步骤(1’)所得产物进行连接反应,纯化回收连接产物;(3’)将步骤(2’)所得连接产物延伸补齐末端,加入测序引物进行pcr扩增,纯化回收,上机测序。优选地,所述分子接头在连接反应体系中的终浓度为45-55μm,45μm、46μm、47μm、48μm、49μm、50μm、51μm、52μm、53μm、54μm或55μm,优选为50μm。与现有技术相比,本发明具有如下有益效果:(1)本发明的分子接头可以用于扩增子建库,通过特异性基因引物延伸从而富集建库;(2)本发明的分子接头包括单分子随机碱基标签区,通过调整5’端引物结合区、单分子标签区和共有连接区的碱基数,解决测序过程中固定错误率导致的留存率过低的问题,无需额外提高测序深度,降低测序成本;(3)本发明提供的分子接头制备方法简单,制备工艺条件宽松,通过一条单链多核苷酸退火后酶切,利用磁珠纯化,降低了接头制备的难度和时间成本。附图说明图1是现有技术的y字形接头,其中a和b代表两个引物结合区,α和α'代表两个互相匹配的兼并序列互补对区;图2是现有技术的y字形接头与待测底物分子连接后的结构示意图,其中β和β’是另外两个互相匹配的简并序列互补配对区;图3是现有技术的y字形接头制备工艺;图4是本发明实施例1的单链多核苷酸退火产物示意图,其中,501为引物结合区,502为单分子标签随机编码区,503为第一互补序列区,504为环形连接区,505为第二互补序列,506为生物素标记;图5是本发明实施例1的退火产物酶切反应示意图,其中507为带有链霉素的磁珠;图6是本发明实施例2的分子接头结构示意图,其中401为5’端引物结合区,402为单分子标签区,403为第一序列,404为第二序列;图7是本发明实施例2的分子接头与待测底物连接反应示意图;图8是本发明实施例2的接头引物和基因特异性引物的工作原理示意图,其中a是5’端引物结合区,a’是a的互补配对区,gsp是基因特异性识别序列,b是通用扩增序列ubr,b’是b的互补配对区,801是il-forward引物,802是spe引物。具体实施方式为更进一步阐述本发明所采取的技术手段及其效果,以下通过具体实施方式来进一步说明本发明的技术方案,但本发明并非局限在实施例范围内。pcr引物序列及扩增子设计数量spe引物序列由基因特异性序列(genespecificprimer,gsp)和通用扩增序列(universalbindingregion,ubr)两部分退火匹配区构成,spe引物设计涵盖人类基因组上的8206个目标扩增区域。结合建库引物实施建库,通过测序的数据质量,回帖率和特定碱基分子标签数量及每个分子标签的深度等参数测试实际接头建库效果,spe引物的序列为5'-ubr-gsp-3';其中ubr序列(seqidno.5):5’-aatgtacagtattgcgttttg-3’;gsp序列由8206个匹配不同基因组区域的序列特异性序列组成,长度范围22-35个碱基,限于篇幅,本实施例所用8206个特异性序列中的六条gsp序列示例性地如seqidno.6-11所示,本领域技术人员可以根据不同的测序目标区域进行特异性序列的设计。gsp序列包括如seqidno.6-11所示的核苷酸序列,具体为:seqidno.6:gctgtagacactattgaagaaaatac;seqidno.7:acctatggacactcagtaaaaac;seqidno.8:cacggtgtagttgatggaccaggagtgaaagttcag;seqidno.9:cgtgtggactctgtgcggtgcc;seqidno.10:gtcacccaggaggtaacctgacacccttg;seqidno.11:gcgtcggactctctgtctagacatcatctgatt;建库通用引物p5(seqidno.12):aatgatacggcgaccaccgacaaaacgcaatactgtacatt;il-forward引物(seqidno.13):gctcttccgatctgtctcgtgggctcggagatgt;通用引物il-p7(seqidno.14):caagcagaagacggcatacgagatcgagaaggctaga;实施例1分子接头制备(1)设计单链多核苷酸的核苷酸序列,由idt公司合成,将合成的单链多核苷酸干粉配制为100μm浓度的溶液,取100μl置于200μl的pcr管;置于pcr仪上,设置热盖为105℃;设置程序为95℃5分钟,设置每分钟降温1℃至1小时;(2)反应1小时后,取90ul至于酶切反应体系中,pcr热盖关闭,37℃反应12小时,酶切体系见表1;(3)反应结束后,加入20μl琏霉素磁珠,移液枪轻轻吹打10次后静置10分钟后,再次吹打5次后静置10分钟。将反应体系至于磁铁架上静置1分钟,小心吸取上清,转移到1.5mlpcr管中,加入500μl-20℃预冷100%酒精,上下倒置10次混匀。恒温超速离心机设置4℃在10000×g重力下离心30分钟;去上清后,加入500μl-20℃预冷75%酒精,移液枪轻轻吹打混匀后,恒温超速离心机4℃10000xg离心30分钟。弃上清,室温超净工作台中倒置干燥10分钟后正置干燥5分钟。加入50μlddh2o稀释后取1μl定量,根据浓度加入适量ddh2o定量成50μm的接头母液止于-80℃预冷75%酒精,移液枪轻轻吹打混匀后,恒温超速离心机4℃10000×g离心30分钟。弃上清,室温超净工作台中倒置干燥10分钟后正置干燥5分钟。加入50μlddh2o稀释后取1μl定量,根据浓度加入适量ddh2o定量成50μm的接头母液置于-80℃长期保存。所述单链多核苷酸的核苷酸序列如seqidno.1所示,具体如下:gtctcgtgggctcggagatgtgctcttccgatctnnnnnnnnnnnntgactgtagaaga-gcttgcagtgggcu*u*acatggcgatagctagact-tcttctacagtca;其中第1-34bp为5’端引物结合区,35-46bp为单分子标签区,47-59bp为第一互补序列区,60-93bp为环形连接区,94-106bp为第二互补序列区。表1酶切反应体系试剂体积/μl终浓度oligo10050μm10×nebcutsmartbuffer201×hpych4iii(5u/μl)200.5u/μlddh2o60-本发明中,ngs建库分子接头的制备方法原理如图4所示,本发明所述接头构建的原料分子为化学方法合成的单链多核苷酸,由5部分构成,分别为引物结合区501,单分子标签随机编码区502,第一互补序列区503,环形连接区504,和第二互补序列区505,特别的,环形连接区的特征为1个或者多个核苷酸上具备生物素标记506。单分子退火后,由于单链自身退火的偏好性,接头原料分子序列会退火形成发卡式二级结构。另外也会存在少量由两条原料分子退火形成的链式结构;限制性酶切反应过程如图5所示,接头原料分子在酶切位点处断裂,形成不带生物素标记的成品接头部分和带有生物素标记的部分;另外未发生酶切反应或者部分酶切的原料分子也带有生物素标记;利用带有琏霉素的磁珠对反应体系内的核酸分子进行负选;酶切下的核酸条带和未发生酶切反应的原料分子都会被去除,留下高纯度的产物接头结构序列,经过定量、沉淀步骤后即得到成品接头产物。实施例2测序实验1、dna片段化起始dna为人源dna,用量为80ng,所需试剂均购自neb,根据表2配制反应体系,向配制好的体系中加入2μldsdna片段化酶,涡旋震荡3秒后置于37℃pcr仪上孵育25分钟后磁珠纯化。表2dna片段化反应体系试剂体积/μldna510×reactionbuffer2ddh2o13total182、修复片段化的dna将片段化的dna稀释成50μl,配制修复反应体系,见表3,将配制好的反应体系置于pcr仪上,热盖70℃,反应条件20℃30分钟,65℃30分钟。表3修复反应体系试剂体积/μldna50nebnextultraiiendprepenzymemix3nebnextultraiiendprepreactionbuffer7total603、连接分子接头连接分子接头的反应体系见表4,将配制好的反应体系20℃孵育15分钟后加入3μluserenzyme37℃孵育15分钟,磁珠纯化回收产物。表4分子接头连接反应体系试剂体积/μlrepaireddna60nebnextultraiiligationmastermix30nebnextligationenhancer1实施例1的分子接头2.5total93.54、特异性扩增步骤将上一步的产物配制为10μl产物溶液,按照表5配制反应液,设置pcr反应程序见表6.表5反应液组成表6pcr反应程序stepduration95℃13min98℃2min98℃15sec6cycles65℃15min72℃5min4℃5min4℃hold5、通用扩增步骤将步骤4得到的反应液磁珠纯化后配制成16μlddh2o,再配制如表7所示反应体系,pcr反应程序见表8。表7表8温度时间95℃13min98℃2min98℃15sec14cycles60℃2min72℃5min4℃hold6、将步骤5所得产物纯化回收后,定量质控,上机测序。7、按照步骤1-6,平行构建3个文库n001,n002和n003,illuminanextseq500双端150bp测序。本发明的分子接头可以用于pcr扩增子建库,如图6所示,接头由5'端的引物结合区401;由随机碱基构成的单分子标签402;以及由第一序列403和第二序列404互补构成的共有连接区域,其中403序列的3'端突出一个粘性末端t(胸腺嘧啶)碱基并羟基化,404序列5'端磷酸化。接头与待测底物片段连接反应如图7所示,接头连接到底物后,在建库过程中404沿5'到3'端的方向延伸补齐末端的突出部分。建库过程中,接头引物ilforward引物(seqidno.13)和spe引物的工作原理如图8所示,图中底物双分子片段标注为top和bottom,引物分为两部分,虚线所示为gsp序列,实线所示为ubr序列(即b),第一轮pcr过后,来自top链的序列和来自bottom链的序列分别由gsp引物和末端引物延伸获得。最终扩增获得带有相同分子标签的来自同一底物片段两条链的序列。带有相同分子标签所有reads都包含的突变位点判定为真正的生物学突变。实验结果处理与分析对上机测序fastq原始数据应用smcounter分析流程进行生物信息分析。主要包括对reads进行trim处理、对reads进行参考基因组回帖、reads比对后过滤、基因特异性引物识别、barcode聚类、基因特异性引物去除、采用开源软件smcounter识别变异位点。测序数据质量见表9.表9由表9可知,三次平建库测序在覆盖率,单个碱基的分子标签数量,每个分子标签的reads深度,回帖率都非常稳定。申请人声明,本发明通过上述实施例来说明本发明的详细方法,但本发明并不局限于上述详细方法,即不意味着本发明必须依赖上述详细方法才能实施。所属
技术领域:
的技术人员应该明了,对本发明的任何改进,对本发明产品各原料的等效替换及辅助成分的添加、具体方式的选择等,均落在本发明的保护范围和公开范围之内。sequencelisting<110>凯杰(苏州)转化医学研究有限公司<120>一种ngs建库分子接头及其制备方法和用途<130>2019<160>14<170>patentinversion3.3<210>1<211>106<212>dna<213>人工合成序列<220><221>misc_feature<222>(35)..(46)<223>nisa,c,g,toru<400>1gtctcgtgggctcggagatgtgctcttccgatctnnnnnnnnnnnntgactgtagaagag60cttgcagtgggcuuacatggcgatagctagacttcttctacagtca106<210>2<211>34<212>dna<213>人工合成序列<400>2gtctcgtgggctcggagatgtgctcttccgatct34<210>3<211>13<212>dna<213>人工合成序列<400>3tgactgtagaaga13<210>4<211>13<212>dna<213>人工合成序列<400>4tcttctacagtca13<210>5<211>21<212>dna<213>人工合成序列<400>5aatgtacagtattgcgttttg21<210>6<211>26<212>dna<213>人工合成序列<400>6gctgtagacactattgaagaaaatac26<210>7<211>23<212>dna<213>人工合成序列<400>7acctatggacactcagtaaaaac23<210>8<211>36<212>dna<213>人工合成序列<400>8cacggtgtagttgatggaccaggagtgaaagttcag36<210>9<211>22<212>dna<213>人工合成序列<400>9cgtgtggactctgtgcggtgcc22<210>10<211>29<212>dna<213>人工合成序列<400>10gtcacccaggaggtaacctgacacccttg29<210>11<211>33<212>dna<213>人工合成序列<400>11gcgtcggactctctgtctagacatcatctgatt33<210>12<211>41<212>dna<213>人工合成序列<400>12aatgatacggcgaccaccgacaaaacgcaatactgtacatt41<210>13<211>34<212>dna<213>人工合成序列<400>13gctcttccgatctgtctcgtgggctcggagatgt34<210>14<211>37<212>dna<213>人工合成序列<400>14caagcagaagacggcatacgagatcgagaaggctaga37当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1