萘酰亚胺或苝酰亚胺荧光标记的O6-苄基鸟嘌呤衍生物及其制备方法与流程
本发明属于蛋白质标记物的合成,涉及萘酰亚胺或苝酰亚胺荧光标记的o6-苄基鸟嘌呤衍生物及其制备方法。
背景技术:
蛋白质特异性的小分子荧光染料标记技术克服了荧光蛋白(fluorescentproteinfp)在蛋白质研究领域的诸多局限性。该类技术通过标准的基因工程手段构建蛋白质与多肽,蛋白质标签的重组蛋白,利用荧光探针配体与相应多肽、蛋白质标签的特异性高效识别,实现对靶蛋白的定点荧光标记。snap-tag凭借对靶蛋白所具有的高特异性和稳定性,以及配体功能多样性等独特优势,从众多靶向标记技术中脱颖而出。成为了最优秀的融合标签之一,并在众多领域得到了广泛应用。比如,活体成像、细胞成像、蛋白质体外研究、蛋白质体内研究、微生物致病机理研究、疾病诊断等。
snap-tag是由johnsson等于2003年开发的,它是通过改造人的o6-鸟嘌呤-dna烷基转移酶(humano6-alkylguanine-dnaalkyitransferase,hagc)获得的一种特异性蛋白质标签。hagt是一种由207种氨基酸组成的单体蛋白,它可以将甲基化dna鸟嘌呤o6位置的的甲基转移到自身的hagt有活性的半胱氨酸巯基(cys145)上,从而实现对dna的修复。针对hagt结构的改造一方面降低原有蛋白结构于甲基化dna的相互作用,另一方面提高其与o6修饰的苄基鸟嘌呤(benzyiguanine,bg)的特异性反应活性。位于snap-tag活性位点的半胱氨酸巯基(cys145)可以与bg的苄基发生反应,在蛋白与底物之间形成非常稳定的共价键实现标记,其过程如下所示:
snap-tag独特的优点有:蛋白标签与底物反应速度快且特异性高;通过共价键结合的蛋白稳定性好,即使在体外分析sds-page的变性条件下仍可稳定标记;通过模块化的设计将标签特异性底物bg与功能染料结合,灵活性高;便于衍生多样化的荧光染料,通用性强,可满足各种荧光成像的需求。目前,snap-tag在细胞内蛋白质标记、体外分析及靶向荧光探测的研究中获得了非常广泛的应用。不过这一方法也存在一定的不足;标签特异性底物bg的合成较复杂,且与染料结合后可能存在溶解分散性较差的问题;大多数染料在使用时,标记前后荧光信号无明显差异,需要充分洗涤去除非特异性染色,可能存在背景干扰。而本发明为改良snap-tag技术中的不足,提供了新型蛋白质特异性的分子荧光标记物。
技术实现要素:
本发明的一个目的是提供一种萘酰亚胺和苝酰亚胺荧光标记的o6-苄基鸟嘌呤衍生物。
本发明的目的之二在于提供该衍生物的制备方法。
为达到上述目的id,本发明采用如下反应机理:
根据上述反应机理,本发明采用如下技术方案:
一种萘酰亚胺或苝酰亚胺荧光标记的o6-苄基鸟嘌呤衍生物,其特征在于该化合物的结构通式为下列之一:
a.
b.
c.
d.
其中r=烷基、环烷基、芳基、杂芳基或聚氧乙烯醚链。
一种根据权利要求1制备所述的萘酰亚胺荧光标记的o6-苄基鸟嘌呤衍生物a的方法,其特征在于该方法的具体步骤为:
a.将6-氯鸟嘌呤和n-甲基吡咯烷按1:1.5-1:2.1的摩尔比溶于n,n-二甲基甲酰胺中,60℃-100℃,反应24h-48h,反应结束抽滤,用丙酮洗涤滤渣,抽滤得到化合物m,其结构式为:
b.将4-溴-1,8-萘酐和有机磺酸盐按1:1.2-1:1.5的摩尔比溶于无水乙醇中,回流条件下反应6h-12h,冷却,滤出固体粗产物,再经分离提纯得到化合物ⅰb,其结构式为:
c.将步骤b所得中间产物ⅰb、氨甲基苄醇和碳酸钾按1:1:1.2-1;1.5:2.0的摩尔比溶于n,n-二甲基甲酰胺,回流反应6h~8h,得到深褐色反应液,调节ph为4~5,除去大部分溶剂,得到糊状物质,抽滤,得到粗产物;再经分离提纯得化合物ⅰc,其结构式为:
d.将步骤c所得中间产物ⅰc和步骤a中所得中间体m和t-buok按1:2:5-1:3:5的摩尔比溶于n,n-二甲基甲酰胺中,惰性气氛保护下,反应3h-8h,经分离提纯得到萘酰亚胺荧光标记的o6-苄基鸟嘌呤衍生物a,其结构式为:
一种制备根据权利要求1所述的萘酰亚胺荧光标记的o6-苄基鸟嘌呤同分子孪生的荧光蛋白质标记物c的方法,特征在于该方法的具体步骤为:
e.将4-溴-1,8-萘酐和氨甲基苄醇按1:1.2-1:1.5的摩尔比溶于无水乙醇中,回流条件下反应6h-12h,冷却,滤出固体粗产物,再经分离提纯得到化合物ⅲa,其结构式为:
f.将步骤e所得中间产物ⅲa、氨甲基苄醇和碳酸钾按1:1:1.2-1;1.5:2.0的摩尔比溶于n,n-二甲基甲酰胺,回流反应6h~8h,得到深褐色反应液,调节ph为4~5,除去大部分溶剂,得到糊状物质,抽滤,得到粗产物;再经分离提纯得化合物ⅲb,其结构式为:
g.将步骤f所得产物ⅲb、步骤a所得产物m、t-buok按1:2:5-1:3:5的摩尔比溶于n,n-二甲基甲酰胺中,惰性气氛保护下,反应3h-8h,分离得到萘酰亚胺荧光标记的o6-苄基鸟嘌呤同分子孪生的荧光蛋白质标记物c,其结构式为:
一种制备根据权利要求1所述的苝酰亚胺荧光标记的o6-苄基鸟嘌呤衍生物的方法,其特征在于该方法的具体步骤为:
a.将四氯苝酐和有机磺酸盐按1:1.2-1:1.5的摩尔比溶于乙二醇甲醚中,100℃-120℃反应
6h-12h,经分离提纯得到化合物ⅱa,其结构式为:
所述的四氯苝酐的结构式为:
所述的有机磺酸盐的结构式为:nh2rso3h;
b.将步骤a所得中间产物ⅱa和氨甲基苄醇按照1:1.2-1:1.5的摩尔比溶于无水乙醇中,回流条件下搅拌反应6h-12h,除去溶剂,所得粗产物经分离提纯得到固体化合物ⅱb,其结构式为:
c.将步骤b所得中间产物ⅱb和对叔丁基酚和碳酸钾按1:4:4-1:5:6的摩尔比溶于n,n-二甲基甲酰胺,回流条件下搅拌反应6~8小时,所得粗产品经分离提纯得到化合物ⅱc,其结构式为:
d.将步骤c所得产物ⅱc、权利要求2步骤a中产物m和t-buok按1:1.2:3-1:2.1:5的摩尔比,惰性气氛保护下,反应3h-5h,经分离提纯得到苝酰亚胺荧光标记的o6-苄基鸟嘌呤衍生物b,其结构式为:
一种制备根据权利要求1制备所述苝酰亚胺荧光标记的o6-苄基鸟嘌呤同分子荧光孪生标记物的制备方法,其特征在于该方法的具体步骤是:
e.将四氯苝酐和氨甲基苄醇按1:2-1:3的摩尔比溶于乙二醇甲醚中,100℃-140℃反应6h-12h,经分离提纯得到化合物ⅳa,其结构式为:
f.将步骤e所得产物ⅳa和对叔丁基酚和碳酸钾按1:4:5-1:5:5的摩尔比溶于n,n-二甲基甲酰胺,回流条件下搅拌反应6~8小时,然后蒸除溶剂,所得粗产品经分离提纯得到化合物ⅳb,其结构式为:
g.将步骤f所得产物ⅳb和权利要求2步骤a中产物m和t-buok按1:2:5-1:2.5:5的摩尔比,惰性气氛保护下,反应3h-5h,经分离提纯得到苝酰亚胺荧光标记的o6-苄基鸟嘌呤同分子荧光孪生标记物d,其结构式为:
本发明的牛磺酸化学修饰的具有优良水溶性的的萘酰亚胺和苝酰亚胺,制备高荧光产率和良好水溶性的o6-苄基鸟嘌呤衍生物,以改进snap-tag蛋白质标记技术中,标签特异性底物o6-苄基鸟嘌呤衍生物(benzylguanine,bg)的水溶性较差和水洗除去非特异性染色背景干扰不足的问题。本发明用牛磺酸与萘酐和苝酐反应,获得萘酰亚胺和苝酰亚胺的磺酸盐,产物呈现良好的水溶性,进一步用氨甲基苄醇进行反应,然后与鸟嘌呤活性中间体偶联,获得目标产物。同时合成目标分子的二聚体(利用同分子孪生协同效应),可望提高该类特异性标记蛋白的靶向性能。
附图说明
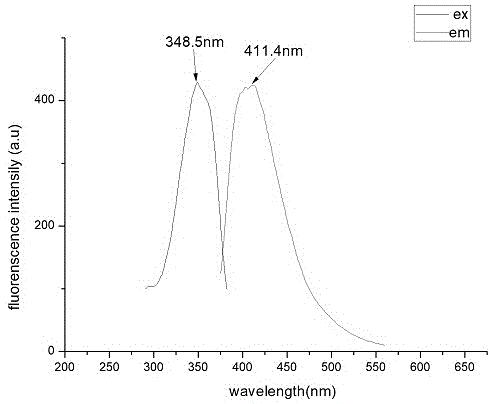
图1是实施例一中萘酰亚胺荧光标记的o6-苄基鸟嘌呤衍生物a1的归一化荧光激发发射光谱;
图2是实施例2中苝酰亚胺荧光标记的o6-苄基鸟嘌呤衍生物c的归一化荧光激发发射光谱。
具体实施方式
本发明结合实施例做进一步说明,但本发明不受下述实施例的限定。
实施例一:萘酰亚胺荧光标记的o6-苄基鸟嘌呤衍生物a1的制备
a.在250ml圆底烧瓶中加入6-氯鸟嘌呤1.69g(10mmol),80mln,n-二甲基甲酰胺作溶剂,加入n-甲基吡咯1.75g(21mmol),60℃搅拌24小时。反应完成后,冷却,沉淀物用20ml丙酮洗涤,然后过滤纯化,得到白色固体1.94g,产率为76.5%。1hnmr(500mhz,dmso-d6,ppm):δ13.41(s,1h),8.35(s,1h),7.12(s,2h),4.67–4.48(m,2h),4.03–3.89(m,2h),3.65(s,3h),2.30–2.19(m,2h),2.06(dd,j=7.4,4.7hz,2h).其结构式为;
b.在50ml圆底烧瓶中加入4-溴-1,8-萘酐277.0mg(1.0mmol),牛磺酸125.1mg(1.0mmol),20mletoh作溶剂,78℃回流搅拌6小时。反应完成后,抽滤得到粗产物,然后经柱层析分离,以二氯甲烷:甲醇=60:1作为洗脱剂,柱层析分离纯化得褐色固体ⅰba;279.6mg,产率为79%。1hnmr(500mhz,d2o,ppm):δ7.83(d,j=7.2hz,2h),7.59(d,j=7.9hz,1h),7.35(t,j=7.9hz,1h),7.26(d,j=7.9hz,1h),4.06(t,j=5.6hz,2h),3.15(t,j=5.7hz,2h).其结构式为;
c.在50ml圆底烧瓶中加入中间体ⅰba355.0mg(1.0mmol),氨甲基苄醇137.0mg(1.0mmol),20mln,n-二甲基甲酰胺作溶剂,加入碳酸钾138.0mg(1mmol),回流搅拌8小时。反应完成后,调节ph为4-5,将溶剂减压蒸馏,剩余物经柱层析分离,以二氯甲烷:甲醇=40:1作为淋洗液,柱层析分离纯化得褐色固体ⅰca264.0mg,产率为60%。1hnmr(500mhz,dmso-d6,ppm):δ8.57(d,j=7.2hz,2h),8.33(d,j=7.9hz,1h),8.20(d,j=7.9hz,1h),7.98(t,j=7.9hz,1h),7.35-7.36(m,1h),7.33(d,j=7.9hz,2h),7.25(d,j=8.0hz,2h),5.22(s,2h),5.14(t,j=5.6hz,1h),4.44(d,j=5.6hz,2h).3.15(t,j=5.6hz,2h),4.06(t,j=5.6hz,2h).其结构式为;
d.在50ml中加入中间体ⅰca440.0mg(1.0mmol),中间体m635.1mg(2.5mmol),10ml干燥的n,n-二甲基甲酰胺,加入t-buok684.80mg(5mmol),在n2保护下常温搅拌3小时。tlc点板跟踪,反应完成后,将溶剂减压蒸馏,调节ph为4-5,剩余物经柱层析分离,以二氯甲烷:甲醇=20:1作为淋洗液,柱层析分离纯化得深棕色固体303.7mg,产率为53%。1hnmr(500mhz,dmso-d6,ppm):δ12.41(s,1h),8.57(d,j=7.2hz,2h),8.44(d,j=7.2hz,1h)8.33(d,j=7.9hz,1h),8.20(d,j=7.9hz,1h),7.98(t,j=7.9hz,1h),7.35(m,1h),7.33(d,j=7.9hz,2h),7.25(d,j=8.0hz,2h),5.36(s,2h),5.22(s,2h),5.14(t,j=5.6hz,1h),4.44(d,j=5.6hz,2h),4.06(t,j=5.6hz,2h),3.15(t,j=5.7hz,2h).其结构式为:
实施例二:苝酰亚胺荧光标记的o6-苄基鸟嘌呤衍生物b1的制备;
a.在50ml圆底烧瓶中加入四氯苝酐532.0mg(1.0mmol),20ml乙二醇甲醚作溶剂,牛磺酸125.1mg(1.0mmol),100℃回流搅拌6小时。反应完成后,抽滤得到粗产物,然后经柱层析分离,以二氯甲烷:甲醇=60:1作为淋洗液,柱层析分离纯化得深红色固体化合物ⅱaa;477.9mg,产率为75%。1hnmr(500mhz,dmso-d6,ppm):δ7.92(s,2h),6.28(s,2h),3.64(t,j=5.6hz,2h),3.33(t,j=5.7hz,2h).其结构式为:
b.在50ml圆底烧瓶中加入中间体ⅱaa637.0mg(1.0mmol),氨甲基苄醇137.0mg(1.0mmol),20mldmf作溶剂,回流搅拌8小时。反应完成后,将溶剂减压蒸馏,剩余物经柱层析分离,以二氯甲烷:甲醇=40:1作为淋洗液,柱层析分离纯化得深紫红色固体ⅱba612.4mg,产率81%。1hnmr(500mhz,dmso-d6,ppm):δ7.92(s,2h),7.33(d,j=7.9hz,2h),7.25(d,j=8.0hz,2h),6.28(s,2h),5.22(s,2h),5.14(t,j=5.7hz,1h),4.44(d,j=5.6hz,2h),3.64(t,j=5.6hz,2h),3.33(t,j=5.6hz,2h).其结构式为:
c.在50ml中加入中间体ⅱba756.0mg(1.0mmol),对叔丁基酚750mg(5mmol),20mln,n-二甲基甲酰胺,加入碳酸钾414.0mg(3mmol),130℃反应6h。tlc点板跟踪,反应完成后,将溶剂减压蒸馏,调节ph为4-5,剩余物经柱层析分离,以二氯甲烷:甲醇=30:1作为淋洗液,柱层析分离纯化得深紫红色固体化合物ⅱca1.0302g,产率85%。1hnmr(500mhz,dmso-d6,ppm):δ7.92(s,2h),7.33(d,j=7.9hz,2h),7.29(d,j=8.5hz,8h),7.25(d,j=8.0hz,2h),6.74(d,j=8.0hz,8h),6.28(s,j=8.5hz,2h),5.22(s,2h),5.14(t,j=5.7hz,1h),4.44(d,j=5.6hz,2h),3.33(t,j=5.7hz,2h,),3.64(t,j=5.6hz,2h),1.42(s,36h).其结构式为:
d.在50ml中加入中间体ⅱca1213.5mg(1.0mmol),中间体m635.1mg(2.5mmol),10ml干燥的n,n-二甲基甲酰胺,加入t-buok684.80mg(5mmol),在n2保护下常温搅拌3小时。tlc点板跟踪,反应完成后,将溶剂减压蒸馏,调节ph为4-5,剩余物经柱层析分离,以二氯甲烷:甲醇=20:1作为淋洗液,柱层析分离纯化得深棕色纯产品化合物874.2mg,产率63%。1hnmr(500mhz,dmso-d6,ppm):δ12.41(s,1h),8.44(d,j=7.2hz,1h),7.92(s,2h),7.33(d,j=7.9hz,2h),7.25(d,j=8.0hz,2h),7.29(d,j=8.5hz,8h),6.74(d,j=8.0hz,8h),6.28(s,2h),5.36(s,2h),5.22(s,2h),4.44(d,j=5.6hz,2h),3.33(t,j=5.7hz,2h),3.64(t,j=5.6hz,2h),1.42(s,36h)
实施例三:萘酰亚胺荧光标记的o6-苄基鸟嘌呤同分子孪生的荧光蛋白质标记物c的方法:
a.在50ml圆底烧瓶中加入4-溴-1,8-萘酐554.0mg(2.0mmol),氨甲基苄醇274.0mg(2.0mmol),20ml无水乙醇作溶剂,78℃回流搅拌6小时。反应完成后,抽滤得到粗产物,然后经柱层析分离,以二氯甲烷:甲醇=100:1作为淋洗液,柱层析分离纯化得白色固体为纯产品化合物ⅲa636.6mg,产率82%。1hnmr(400mhz,dmso-d6,ppm):δ8.57(d,j=7.2hz,2h),8.33(d,j=7.9hz,1h),8.20(d,j=7.9hz,1h),7.98(t,j=7.9hz,1h),7.33(d,j=7.9hz,2h),7.25(d,j=8.0hz,2h),5.22(s,2h),5.14(t,j=5.7hz,1h),4.44(d,j=5.6hz,2h).其结构式为:
在50ml圆底烧瓶中加入中间体ⅲa395.0mg(1.0mmol),氨甲基苄醇137.0mg(1.0mmol),20mln,n-二甲基甲酰胺作溶剂,加入碳酸钾138.0mg(1mmol),回流搅拌8小时。反应完成后,调节ph为4-5,将溶剂减压蒸馏,剩余物经柱层析分离,以二氯甲烷:甲醇=40:1作为淋洗液,柱层析分离纯化得浅褐色固体ⅲb327.6mg,产率70%。1hnmr(400mhz,dmso-d6,ppm):δ8.57(d,j=7.2hz,1h),8.53(d,j=8.5hz,1h),8.33(d,j=7.9hz,1h),8.20(d,j=7.9hz,1h),7.98(t,j=7.9hz,1h),7.35-7.36(m,1h),7.33(d,j=7.9hz,4h),7.25(d,j=8.0hz,4h),5.22(s,4h),5.14(t,j=5.7hz,2h),4.44(d,j=5.6hz,4h).其结构式为:
在50ml中加入中间体ⅰca440.0mg(1.0mmol),中间体m1016mg(4mmol),10ml干燥的dmf,加入t-buok1088mg(8mmol),在n2保护下常温搅拌3小时。tlc点板跟踪,反应完成后,将溶剂减压蒸馏,调节ph为4-5,剩余物经柱层析分离,以二氯甲烷:甲醇=20:1作为淋洗液,柱层析分离纯化得深棕色纯产品化合物574.4mg,产率80%。1hnmr(500mhz,dmso-d6,ppm):δ12.41(s,2h),8.57(d,j=7.2hz,2h),8.44(d,j=7.2hz,2h)8.33(d,j=7.9hz,1h),8.20(d,j=7.9hz,1h),7.98(t,j=7.9hz,1h),7.35-7.36(m,1h),7.33(d,j=7.9hz,4h),7.25(d,j=8.0hz,4h),5.36(s,4h),5.22(s,4h),4.44(d,j=5.6hz,4h).其结构式为:
实施例四:苝酰亚胺荧光标记的o6-苄基鸟嘌呤同分子荧光孪生标记物d的制备:
a.在50ml圆底烧瓶中加入四氯苝酐532.0mg(1.0mmol),20ml无水乙醇作溶剂,氨甲基苄醇274.0mg(2.0mmol),100℃回流搅拌6小时。反应完成后,抽滤得到粗产物,然后经柱层析分离,以石油醚:乙酸乙酯=1:1作为淋洗液,柱层析分离纯化得深红色固体化合物ⅳa477.9mg,产率75%。1hnmr(500mhz,dmso-d6,ppm):δ7.92(s,j=8.5hz,4h),7.33(d,j=7.9hz,4h),7.25(d,j=8.0hz,4h),5.22(s,4h),5.14(t,j=5.7hz,2h),4.44(d,j=5.6hz,4h).其结构式为:
在25ml圆底烧瓶中加入中间体ⅳa230.4mg(0.3mmol),对叔丁基酚225mg(1.5mmol),10mln,n-二甲基甲酰胺,加入碳酸钾124.2mg(0.9mmol),130℃反应6h。tlc点板跟踪,反应完成后,将溶剂减压蒸馏,调节ph为4-5,剩余物经柱层析分离,以二氯甲烷:甲醇=30:1作为淋洗液,柱层析分离纯化得深紫红色固体ⅱca293.8mg,产率80%。1hnmr(500mhz,dmso-d6,ppm):δ7.92(s,4h),7.29(d,j=8.5hz,8h),7.33(d,j=7.9hz,4h),6.74(d,j=8.0hz,8h),7.25(d,j=8.0hz,4h),5.22(s,4h),5.14(t,j=5.7hz,2h),4.44(d,j=5.6hz,4h),1.42(s,36h).其结构式为:
在50ml中加入中间体ⅳb244.8mg(0.2mmol),中间体m203.2mg(0.8mmol),10ml干燥的n,n-二甲基甲酰胺,加入t-buok217.6mg(1.6mmol),在n2保护下常温搅拌3小时。tlc点板跟踪,反应完成后,将溶剂减压蒸馏,剩余物经柱层析分离,以二氯甲烷:甲醇=20:1作为淋洗液,柱层析分离纯化得深紫色纯产品化合物232.2mg,产率78%。1hnmr(500mhz,dmso-d6,ppm):δ12.41(s,2h),8.44(d,j=7.2hz,2h),7.92(s,4h),7.29(d,j=8.5hz,8h),7.33(d,j=7.9hz,4h),6.74(d,j=8.0hz,8h),7.25(d,j=8.0hz,4h),5.36(s,4h),5.22(s,4h),4.44(d,j=5.6hz,4h),1.42(s,36h).其结构式为:
- 还没有人留言评论。精彩留言会获得点赞!