一种二噻吩并苯二酰亚胺衍生物及其制备方法与应用与流程
本发明属于光电材料技术领域,特别涉及一种二噻吩并苯二酰亚胺衍生物及其制备方法与应用。
背景技术:
有机共轭材料可广泛用于光电领域。有机共轭材料的合成目前大部分依赖于两个sp2碳之间有效的c-c键形成。为此,人们发展了多种过渡金属催化的交叉偶联反应,如stille偶联、suzuki偶联(钯催化)以及kumada偶联(镍催化)等,用于杂芳基卤化物与有机金属杂芳烃的偶联。尽管这些的偶联方法对于大多数的π-共轭体系是有效的,但它们通常需要使用活泼的有机金属试剂(例如丁基锂)来制备中间体,往往包含不稳定的、高毒性的或较为繁琐的制备过程来完成c-c键偶联。
近年来,直接c-h芳基化(directc-harlytion,c-h活化)作为一种新兴的c-c键偶联合成方法,受到人们的广泛关注。它可以方便地将芳基卤与非取代的杂芳基直接偶合,而不需要预活化sp2c-h键。这样,可以通过直接c-h键芳基化反应以更少的合成步骤合成得到各种共轭结构小分子或聚合物材料体系;而不涉及高度危险的活泼有机化学试剂,如高活性的丁基锂或高毒性的有机锡等化学试剂。尽管直接c-h芳基化的化学机理得到了广泛研究,但是其在构筑π共轭结构的有机光电材料方面的应用却依然非常有限。
由于c-h键的键能大于c-b键和c-sn键,所以c-h键的直接活化需要能量大、条件较为苛刻;如,一般需要有引导基团来达到固定位置的脱氢。2006年,marclafrance等,使用特戊酸(pivoh)/碳酸钾(k2co3)作为具有引导性质的酸添加剂,组成钯–特戊酸(pivoh)共催化体系,特戊酸根阴离子作为使c-h键断裂的关键部分,降低了c-h断裂的能量,并加速质子从芳基上转移到相对应的碳酸盐。特戊酸共催化体系既可用于富电子的基团也可用于缺电子的基团,使芳基卤与非取代的杂芳c-h活化偶联的产率得到了很大提高。
这类共催化体系成功地应用在一些小分子π共轭有机光电材料体系,包括苯噻唑(bt)衍生物、吡咯吡咯二酮衍生物、萘酰亚二胺(ndi)系列衍生物、噻吩酰胺(tiig)系列衍生物等。并非所有的小分子π共轭有机光电材料体系都适合使用c-h活化的方式偶联,这限制了其在π共轭有机光电材料制备方面的更广泛应用。
通过c-h活化合成π共轭有机光电材料,制备过程简单、适合于规模化量产。存在的主要问题是,缺乏能高效的适用于c-h活化合成π共轭有机光电材料的结构基元。
技术实现要素:
发明目的:为了解决现有技术中的问题,本发明提供了一种二噻吩并苯二酰亚胺衍生物及其制备方法与应用,二噻吩并苯二酰亚胺结构基元可以采用直接c-h活化偶联反应,得到一系列二噻吩并苯二酰亚胺衍生物。该衍生物可以通过使用直接c-h活化偶联反应的方式合成,原料廉价、合成简便,有利于规模化量产;而且该衍生物光谱可调范围宽、能级结构可调控性好,在合成制备π共轭有机光电材料领域有很大应用潜力。
技术方案:为解决上述问题,本发明采用以下技术方案:
一种二噻吩并苯二酰亚胺(dti)衍生物材料,该衍生物以二噻吩并苯二酰亚胺为基本骨架,具有如下式i所示的通式结构:
其中,r为氢、直链烷基、支链烷基、烷氧或芳基中的一种;ar1、ar2为芳香基团;o为氧原子,n为氮原子,s为硫原子。
进一步地,所述芳基ar1、ar2选自下式ii基团中的一种:
其中,r1、r2和r3为c1~c30的直链或支链烷基或烷氧基;*为连接位置;n是氮原子;s是硫原子;o是氧原子。
一种二噻吩并苯二酰亚胺衍生物的制备方法,包括以下步骤:
将二噻吩并苯二酰亚胺、溴取代ar1基团、特戊酸(pivoh)、碳酸盐、催化剂和膦配体溶解在有机溶剂中,加热115~120℃搅拌10~24h,然后加入溶解在有机溶剂中的溴取代ar2基团,加热115~120℃搅拌10~24h反应结束后,分离提纯获得二噻吩并苯二酰亚胺衍生物。
优选地,所述碳酸盐选自k2co3、cs2co3、na2co3、caco3、nahco3中的一种。
优选地,所述催化剂选自pd(pph3)4、pd2(dba)3、双(三苯基膦)醋酸钯、双(三苯基膦)二氯化钯中的一种。
优选地,所述膦配体选自p(2-meoph)3、双(2-二苯基膦乙基)苯基磷、(1-戊基)三苯基溴化磷、双(3,5-二甲苯基)磷中的一种。
优选地,所述有机溶剂选自甲苯、邻二甲苯、三甲苯、二氯苯、四氢呋喃中的至少一种。
优选地,所述二噻吩并苯二酰亚胺、溴取代ar1基团、溴取代ar2基团、pivoh、碳酸盐、催化剂和膦配体的摩尔比为1:0.7~1.8:0.7~1.8:1~1.5:3~4:0.01~0.05:0.08~0.12。
一种二噻吩并苯二酰亚胺衍生物的应用,二噻吩并苯二酰亚胺衍生物可以作为活性层材料或添加组分应用于传感器、电致发光器件、有机太阳能电池或有机场效应晶体管方面,二噻吩并苯二酰亚胺衍生物还可以作为电致变色材料、光致变色材料、传感材料、空穴传输材料、三阶非线性光学材料、防伪材料或伪装材料应用于有机光电领域。
有益效果:与现有技术相比,本发明具有以下有益效果:
(1)二噻吩并苯二酰亚胺结构基元目前尚未见报道,特别是尚未有相关的材料体系应用于有机光电材料领域,而且其c-h活化方式偶联的产率还能进一步提高,光谱可调范围广,能级排列调节性好,在光电领域具有潜在应用。
(2)二噻吩并苯二酰亚胺c-h活化合成方法简单,原料易得、条件便利,不需要制备硼酸、硼酸酯或者烷基锡等前驱体,无毒绿色有利于规模化量产。
(3)二噻吩并苯二酰亚胺衍生物的偶联单体选择范围广宽、产率高。
附图说明
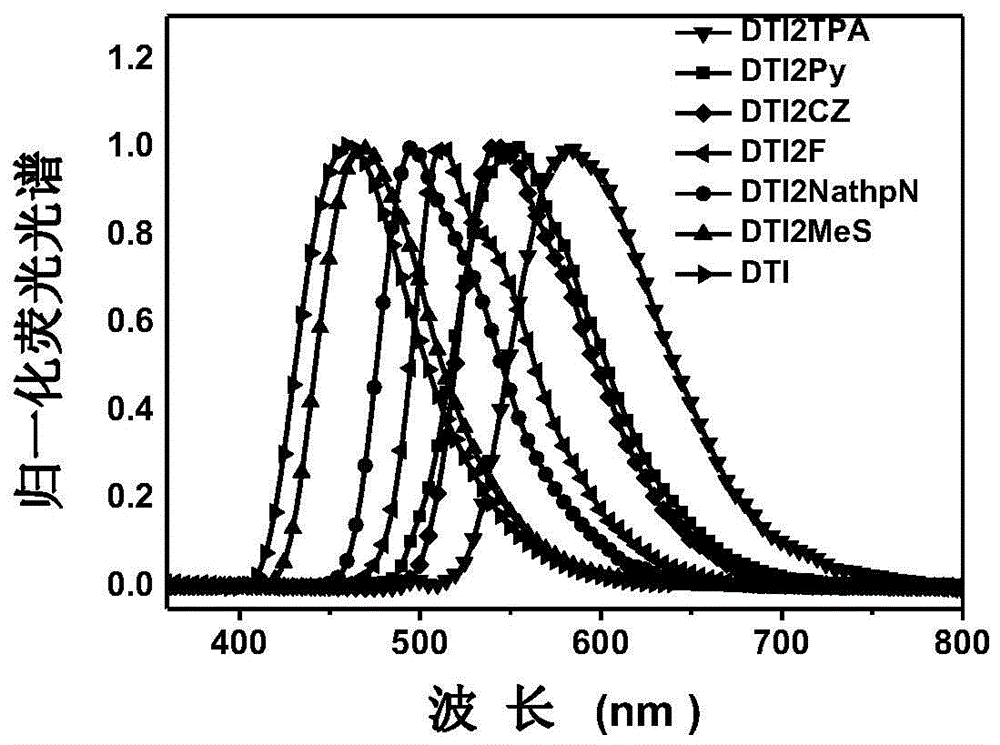
图1是二噻吩并苯二酰亚胺四氢呋喃溶液的荧光光谱;
图2是二噻吩并苯二酰亚胺薄膜的荧光光谱;
图3是二噻吩并苯二酰亚胺的1h核磁共振谱图;
图4是二噻吩并苯二酰亚胺的13c核磁共振谱图;
图5是二噻吩并苯二酰亚胺衍生物的1h核磁共振谱图。
具体实施方式
下面结合实施例对本发明作更进一步的说明。
实施例1
反应路线
根据反应路线,将dti(0.2mmol,84.6mg,1.0eq.),4-溴芘(0..24mmol,1.2eq.),pivoh(0.2mmol,20mg,1.0eq.),配体p(2-meoph)3(0.84mg,0.12eq.)和cs2co3(197mg,3.0eq.)加入25ml克莱氏反应管中,通氮气15min;保证氮气氛围,加入催化剂pd2(dba)3(5.4mg,0.03eq.),再次通氮气15min,保证氮气氛围,注入无水o-xlyene,120℃加热搅拌1天。将溴芘(0.24mmol1.2eq.)溶于0.5mlthf注入到反应瓶,120℃加热搅拌1天。待冷却到室温,使用5~10ml二氯甲烷溶解,用饱和nacl洗3次,然后加无水硫酸镁干燥,过滤后旋干,以石油醚/二氯甲烷为洗脱剂,通过层析硅胶提纯,在dcm/甲醇中重结晶,得到目标化合物t1。
附图3为二噻吩并苯二酰亚胺的核磁共振氢谱谱图,通过氢谱的化学位移确认碳氢活性位的位置,并且裂双峰表明碳氢的活性位是对称存在的氢原子。
附图4为二噻吩并苯二酰亚胺的核磁共振碳谱谱图,通过化学位移峰的个数,确认结构中碳原子的个数复合二噻吩并苯二酰亚胺中的结构。
实施例2
反应路线
根据反应路线,将dti(0.2mmol,84.6mg,1.0eq.),4-溴1,8萘酐(0.24mmol1.2eq.),pivoh(0.2mmol,20mg,1.0eq.),配体p(2-meoph)3(0.84mg,0.12eq.)和cs2co3(197mg,3.0eq.)加入25ml克莱氏反应管中,通氮气15min,保证氮气氛围,加入催化剂pd2(dba)3(5.4mg,0.03eq.),再次通氮气15min,保证氮气氛围,注入无水o-xlyene,120℃加热搅拌1天。将4-溴-1,8-萘酐(0.24mmol1.2eq.)溶于0.5mlthf注入到反应瓶,120℃加热搅拌1天。冷却到室温,使用5~10ml二氯甲烷溶解,用饱和nacl洗3次,然后加无水硫酸镁干燥,过滤后旋干,以石油醚/二氯甲烷为洗脱剂,通过层析硅胶提纯,在dcm/甲醇中重结晶,得到目标化合物t2。
图5为目标产物的核磁共振氢谱谱图,化学位移在7.8ppm和8.7ppm的峰的裂峰状况表明了1,8-萘酐连接在噻吩并苯二酰亚胺的位置是对称的。
实施例3
反应路线
根据反应路线,将dti(0.2mmol,84.6mg,1.0eq.),1-溴-2,4,6三甲苯(0.3mmol3.0eq.),pivoh(0.2mmol,20mg,1.0eq.),配体p(2-meoph)3(0.84mg,0.12eq.)和cs2co3(197mg,3.0eq.)加入25ml克莱氏反应管中,通氮气15min,保证氮气氛围,加入催化剂pd2(dba)3(5.4mg,0.03eq.),再次通氮气15min,保证氮气氛围,注入无水o-xlyene,120℃加热搅拌1天。将1-溴-2,4,6三甲苯(0.3mmol1.2eq.)溶于0.5mlthf注入到反应瓶,120℃加热搅拌1天。待冷却到室温,使用5~10ml二氯甲烷溶解,用饱和nacl洗3次,然后加无水硫酸镁干燥,过滤后旋干,以石油醚/二氯甲烷为洗脱剂,通过层析硅胶提纯,在dcm/甲醇中重结晶,得到目标化合物t3。
实施例4
反应路线
根据反应路线,将dti(0.2mmol,84.6mg,1.0eq.),1-溴-2,4,6三甲苯(0.3mmol3.0eq.),pivoh(0.2mmol,20mg,1.0eq.),配体p(2-meoph)3(0.84mg,0.12eq.)和cs2co3(197mg,3.0eq.)加入25ml克莱氏反应管中,通氮气15min,保证氮气氛围,加入催化剂pd2(dba)3(5.4mg,0.03eq.),再次通氮气15min,保证氮气氛围,注入无水o-xlyene,120℃加热搅拌1天。将溶于0.5mlthf的1-溴-2,4,6三甲苯(0.3mmol1.2eq.)注入到反应瓶,120℃加热搅拌1天。待冷却到室温,使用5~10ml二氯甲烷溶解,用饱和nacl洗3次,然后加无水硫酸镁干燥,过滤后旋干,以石油醚/二氯甲烷为洗脱剂,通过层析硅胶提纯,在dcm/甲醇中重结晶,得到目标化合物t4。
实施例5
反应路线
根据反应路线,将dti(0.2mmol,84.6mg,1.0eq.),对甲氧基溴苯(0.6mmol3.0eq.),pivoh(0.2mmol,20mg,1.0eq.),配体p(2-meoph)3(0.84mg,0.12eq.)和cs2co3(197mg,3.0eq.)加入25ml克莱氏反应管中,通氮气15min,保证氮气氛围,加入催化剂pd2(dba)3(5.4mg,0.03eq.),再次通氮气15min,保证氮气氛围,注入无水o-xlyene,120℃加热搅拌1天。待冷却到室温,使用5~10ml二氯甲烷溶解,用饱和nacl洗3次,然后加无水硫酸镁干燥,过滤后旋干,以石油醚/二氯甲烷为洗脱剂,通过层析硅胶提纯,在dcm/甲醇中重结晶,得到目标化合物t5。
实施例6
反应路线
根据反应路线,将dti(0.2mmol,84.6mg,1.0eq.),1-溴-2,4,6三甲苯(0.24mmol1.2eq.),pivoh(0.2mmol,20mg,1.0eq.),配体p(2-meoph)3(0.84mg,0.12eq.)和cs2co3(197mg,3.0eq.)加入25ml克莱氏反应管中,通氮气15min,保证氮气氛围,加入催化剂pd2(dba)3(5.4mg,0.03eq.),再次通氮气15min,保证氮气氛围,注入无水o-xlyene,120℃加热搅拌1天。将2-溴-5甲基-3-氟噻吩(0.24mmol1.2eq.)溶于0.5mlthf注入到反应瓶,在120℃条件下加热搅拌1天。待冷却到室温,使用5~10ml二氯甲烷溶解,用饱和nacl洗3次,然后加无水硫酸镁干燥,过滤后旋干,以石油醚/二氯甲烷为洗脱剂,通过层析硅胶提纯,在dcm/甲醇中重结晶,得到目标化合物t6。
实施例7
反应路线
根据反应路线,将dti(0.2mmol,84.6mg,1.0eq.),4-溴三苯胺(0.24mmol1.2eq.),pivoh(0.2mmol,20mg,1.0eq.),配体p(2-meoph)3(0.84mg,0.12eq.)和cs2co3(197mg,3.0eq.)加入25ml克莱氏反应管中,通氮气15min,保证氮气氛围,加入催化剂pd2(dba)3(5.4mg,0.03eq.),再次通氮气15min,保证氮气氛围,注入无水o-xlyene,120℃加热搅拌1天。将2-溴-9,9-二已基芴(0.24mmol1.2eq.)溶于0.5mlthf注入到反应瓶,120℃加热搅拌1天。待冷却到室温,使用5~10ml二氯甲烷溶解,用饱和nacl洗3次,然后加无水硫酸镁干燥,过滤后旋干,以石油醚/二氯甲烷为洗脱剂,通过层析硅胶提纯,在dcm/甲醇中重结晶,得到目标化合物t7。
对于附图1,六种二噻吩并苯二酰亚胺衍生物的溶液态荧光光谱,光谱峰值范围从470nm到590nm覆盖可见光范围,表明通过对于连接基团ar1、ar2的调节,可以实现非常宽的荧光光谱调节范围。
对于图2,六种二噻吩并苯二酰亚胺衍生物的薄膜态荧光光谱,相对于溶液态的荧光光谱,发射峰红移10~40nm,说明堆积情况不同,对于应用于器件时的效果会有差异。
对于图3,二噻吩并苯二酰亚胺的1hnmr各个氢原子的化学位移,符合二噻吩并苯二酰亚胺的结构。
对于图4,二噻吩并苯二酰亚胺的13hnmr各个氢原子的化学位移,符合二噻吩并苯二酰亚胺的结构。
对于图5,一种二噻吩并苯二酰亚胺的衍生物的1hnmr各个氢原子的化学位移,符合一种二噻吩并苯二酰亚胺衍生物的结构。
以上所述仅是本发明的优选实施方式,应当指出:对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!