一种超高分子量聚(4-烷氧基苯乙烯)及其制备方法与流程
本发明属于烯烃催化聚合技术领域,具体涉及一种超高分子量聚(4-烷氧基苯乙烯)及其制备方法。
背景技术:
4-烷氧基类苯乙烯是一种带有极性烷氧基的单体,通过这种单体直接聚合可以制备得到功能性的聚苯乙烯。这类聚合物除了保持聚苯乙烯优异的机械性能、介电性能、表面性能、阻隔性能、耐溶剂性能和耐热性能外,而且烷氧基赋予了其更好的亲水性和粘附性。4-烷氧基苯乙烯从聚合机理上而言,能够通过自由基、阳离子或者配位聚合。由于烷氧基的给电子效应,4-烷氧基苯乙烯的自由基聚合的聚合活性比较低,所得到的聚(4-烷氧基苯乙烯)分子量通常都很低(小于5万),并且聚合物的分散系数比较宽。4-烷氧基苯乙烯也容易发生阳离子聚合,其单体的转化率较高,然而阳离子聚合的反应调节苛刻,非常容易发生各种副反应,很难获得高分子量聚合物(小于10万),聚合物分子量分布也比较宽。金属催化4-烷氧基苯乙烯的配位聚合也能制备出聚(4-烷氧基苯乙烯)。钛基催化剂能够能催化4-甲氧基苯乙烯制备得到间规的或者无规的产物,稀土催化剂能够制备出高间规的聚(4-甲氧基苯乙烯)(间规度>99%),钯基催化剂也能催化4-甲氧基苯乙烯聚合,制备出无规的聚(4-甲氧基苯乙烯)。然而,由于存在极性甲氧基和金属中心的作用,这些催化剂体系催化4-烷氧基苯乙烯配位聚合的活性通常不高,难以制备得到窄分布、超高分子量的聚(4-甲氧基苯乙烯),所得到的聚合物分子量都在20万以下。
技术实现要素:
本发明的目的在于克服现有技术中聚(4-甲氧基苯乙烯)的分子量较低的缺陷或不足,提供一种超高分子量聚(4-烷氧基苯乙烯)。本发明提供的超高分子量聚(4-烷氧基苯乙烯)的重均分子量超过100万,具有更好的机械性能、热稳定性能和亲水性能。
本发明的另一目的在于提供一种α-二亚胺钯配合物。
本发明的另一目的在于提供一种钯催化剂。
本发明的另一目的在于提供上述超高分子量聚(4-烷氧基苯乙烯)的制备方法。
为实现上述发明目的,本发明采用如下技术方案:
一种超高分子量聚(4-烷氧基苯乙烯),具有式(i)所示结构:
其中,r为甲基或乙基;所述超高分子量聚(4-烷氧基苯乙烯)的重均分子量不低于100万。
本发明提供一种超高分子量聚(4-烷氧基苯乙烯),其重均分子量不低于100万,与商业的聚苯乙烯具有更好的机械性能、热稳定性能和亲水性能。
优选地,所述超高分子量聚(4-烷氧基苯乙烯)的重均分子量为964~4598万。
优选地,所述超高分子量聚(4-烷氧基苯乙烯)的分散系数不大于1.3。
优选地,所述超高分子量聚(4-烷氧基苯乙烯)的分散系数为1.15~1.30。
本发明请求保护一种α-二亚胺钯配合物,具有如式(ii)所示结构:
其中,r1为氢、甲基或异丙基;r2为氢或甲氧基;r3为氢或甲氧基。
本发明研究发现,具有二苯并桶烯骨架的α-二亚胺钯配合物可实现超高分子量(不低于100万)、低分散系数(不高于1.3)聚(4-烷氧基苯乙烯)的制备。一方面,二苯并桶烯骨架的苯环具有吸电子的共轭效应,能够增强钯金属中心的亲电性,提高富电子的4-烷氧基苯乙烯单体的配位插入速率(链增长速率);另一方面,大位阻刚性的二苯并桶烯骨架显示出明显的位阻效应,能够屏蔽钯金属背面空间以及轴向的空间,显著抑制聚合反应中链转移的发生。因而通过提高单体的配位插入速率和降低链转移速率,来制备得到高分子量、窄分布的聚(4-烷氧基苯乙烯)。
优选地,r1、r2和r3均为氢。
优选地,r1为甲基、r2和r3均为氢。
优选地,或r1为异丙基、r2和r3均为氢。
优选地,r1和r2均为氢,r3为甲氧基。
优选地,r1和r3均为氢,r2为甲氧基。
优选地,r1为氢,r2和r3均为甲氧基。
上述α-二亚胺钯配合物的制备方法,包括如下步骤:
s1:蒽和碳酸亚乙烯酯进行d-a加成反应后,再与三氟乙肝酸进行swern氧化反应得二酮
s2:二酮与取代苯胺
s3:配体l与pd(cod)ch3cl反应即得所述α-二亚胺钯配合物。
本发明还请求保护一种钯催化剂,由上述α-二亚胺钯配合物和活化剂组成;所述活化剂为四(3,5-二(三氟甲基)苯基)硼酸钠nabarf、六氟磷酸银agpf6、六氟锑酸银agsbf6中的一种或几种;所述活化剂和α-二亚胺钯配合物的摩尔比为1~5:1。
具体地,活化剂的结构式如下:
上述超高分子量聚(4-烷氧基苯乙烯)的制备方法,包括如下步骤:以4-烷氧基苯乙烯为单体,以权利要求7所述钯催化剂为催化剂,在溶剂中进行聚合反应,即得所述超高分子量聚(4-烷氧基苯乙烯)。
优选地,所述4-烷氧基苯乙烯为4-甲氧基苯乙烯或4-乙氧基苯乙烯。
优选地,所述聚合反应的温度为0~80℃。
优选地,所述溶剂为二氯甲烷、1,2-二氯乙烷、氯仿、氯苯或甲苯中的一种或几种。
优选地,所述单体和钯催化剂的摩尔比为750~10000:1。
与现有技术相比,本发明具有如下有益效果:
(1)本发明提供的超高分子量聚(4-烷氧基苯乙烯)的重均分子量超过100万,具有更好的机械性能、热稳定性能和亲水性能。
(2)本发明提供的α-二亚胺钯配合物及钯催化剂具有优异的催化性能,可制备得到高分子量、窄分布的聚(4-烷氧基苯乙烯)。
(3)本发明提供的制备方法聚合反应条件温和,聚合活性高,单体转化率高,整个聚合反应高效可控。
附图说明
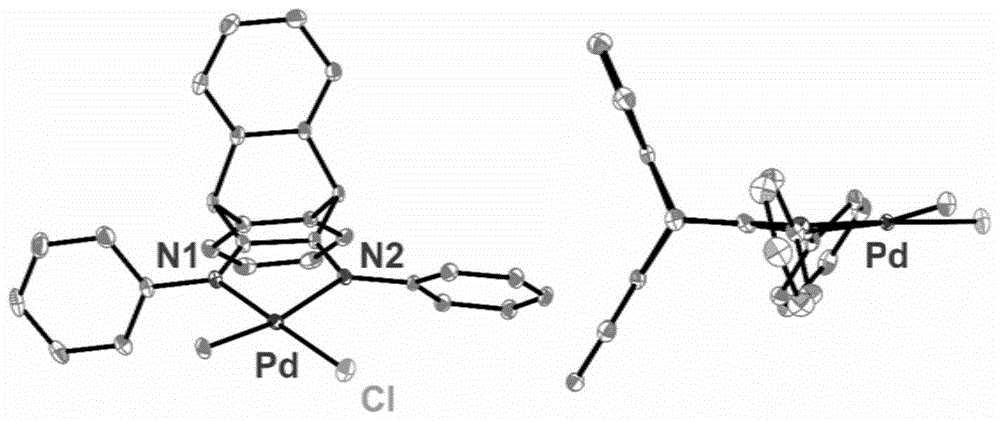
图1为本发明实施例1制备的α-二亚胺钯配合物1的单晶结构示意图;
图2为本发明实施例9和实施例15所制备的聚(4-甲氧基苯乙烯)和聚(4-乙氧基苯乙烯)的核磁氢谱图;
图3为本发明实施例9和实施例15所制备的聚(4-甲氧基苯乙烯)和聚(4-乙氧基苯乙烯)的核磁碳谱图;
图4为本发明实施例1~6制备α-二亚胺钯配合物1~6的反应路线图;
图5为聚(4-甲氧基苯乙烯)的样品图片以及接触角。
具体实施方式
下面结合实施例进一步阐述本发明。这些实施例仅用于说明本发明而不用于限制本发明的范围。下例实施例中未注明具体条件的实验方法,通常按照本领域常规条件或按照制造厂商建议的条件;所使用的原料、试剂等,如无特殊说明,均为可从常规市场等商业途径得到的原料和试剂。本领域的技术人员在本发明的基础上所做的任何非实质性的变化及替换均属于本发明所要求保护的范围。
各实施例中的α-二亚胺钯配合物的结构式如下:
r1为氢、甲基或异丙基;r2为氢或甲氧基;r3为氢或甲氧基,具体地:
α-二亚胺钯配合物1,r1、r2、r3为氢;
α-二亚胺钯配合物2,r1为甲基、r2和r3为氢;
α-二亚胺钯配合物3,r1为异丙基、r2和r3为氢;
α-二亚胺钯配合物4,r1和r2为氢,r3为甲氧基;
α-二亚胺钯配合物5,r1和r3为氢,r2为甲氧基;
α-二亚胺钯配合物6,r1为氢,r2和r3为甲氧基。
实施例1
本实施例提供一种α-二亚胺钯配合物1的合成。其反应路线图如图4,具体如下。
二酮的合成:在氮气的保护下,在250ml高压反应瓶中加入蒽(8.90g,50.0mmol,1eq.)和碳酸亚乙烯酯(31.6ml,500.0mmol,10eq.)加热至180℃反应。8小时后,停止加热,将反应体系冷却至室温,加入150ml甲醇。反应体系洗出大量固体,过滤,固体用甲醇洗涤,真空干燥得到灰色粉末。之后把这个灰色粉末(5.60g,21.0mmol,1eq.)溶解在50ml无水乙醇中,搅拌下缓慢加入氢氧化钾(3.00g,53.5mmol,2.6eq.),继续搅拌6小时。所得混合溶液过滤出去悬浮物,缓慢滴加到水中,析出大量固体。过滤得到白色晶体,继续用水洗涤,真空干燥得到灰白色粉末。最后在氮气的保护下,在250ml的支口烧瓶中,加入30ml二氯甲烷和二甲基亚砜(0.96ml,13.58ml,3.2eq.),在-60℃下搅拌混合。缓慢逐滴滴加三氟乙酸酐(1.73ml,12.26mmol,2.9eq.),搅拌反应体系澄清。上述的灰白色粉末(1.00g,4.27mmol,1eq.)溶解在30ml二氯甲烷中,缓慢逐滴滴加到反应溶液中。1.5小时后,缓慢逐滴滴加三乙胺(3.95ml,26.58mmol,6.6eq.)到反应溶液中,继续搅拌1.5小时后,反应体系温度上升至5℃,将反应溶液倒入150ml的饱和盐酸溶液中,再用二氯甲烷(3×30ml)萃取。合并有机相,用150ml去离子水洗涤,再用无水硫酸镁干燥,过滤,旋转蒸发除去溶剂。固体用甲苯重结晶,得到黄色晶体产物二酮,产率80%。1hnmr(400mhz,cdcl3):7.52-7.45(m,4h,ar-h),7.39-7.34(m,4h,ar-h),5.00(s,2h,ch)13cnmr(400mhz,cdcl3),δ(ppm):153.78,134.86,129.43,126.35,77.27,77.06,76.85。
配体l1的合成:氮气气氛下,上述步骤制备的二酮(2.34g,10.0mmol,1eq)与苯胺(2.04ml,22.0mmol,2.2eq)溶解于100ml甲苯中,加入对甲苯磺酸,加热分水回流24小时。反应后的混合物旋转蒸发除去溶剂,所得固体用乙醇重结晶得到配体l1,产率90%。1hnmr(400mhz,cd3od),δ(ppm):7.36-7.21(m,8h,ar-h),7.03(m,6h,ar-h),6.91(d,4h,ar-h),5.52(s,2h,ch).13cnmr(100mhz,cdcl3),δ(ppm):160.09(c=n),150.41(ar-c-n),138.79(ar-cinbackbone),129.10(ar-c),128.10(ar-c),125.13(ar-cinbackbone),124.58(ar-cinbackbone),120.12(ar-c),50.39(ch).anal.calcdforc28h20n2:c,87.47;h,5.24;n,7.29。
α-二亚胺钯配合物1的合成:α-二亚胺甲基氯化钯配合物1由α-二亚胺配体l1与pd(cod)ch3cl反应得到。氮气氛围下,α-二亚胺配体(1.1mmol)与pd(cod)ch3cl(1.0mmol)加入到预先经高温烘烤除水的schlenk瓶中,再加入无水二氯甲烷(20ml),室温下避光搅拌反应过夜。反应后的溶液经g4过滤球过滤后,减压蒸发浓缩至剩余5ml,再加入无水正己烷(50ml),析出固体。过滤后,固体用无水正己烷洗涤(3×5ml),真空干燥后得到粉末产物。产物为橙红色粉末,反应产率87.2%。1hnmr(400mhz,cdcl3),δ(ppm):7.62-7.50(m,4h,ar-h),7.46-7.28(m,8h,ar-hinbackbone),7.27-7.23(m,2h,ar-h),7.17-7.10(d,2h,ar-h),7.01-6.95(d,2h,ar-h),5.38(s,1h,ch),5.06(s,1h,ch),0.67(s,3h,pd-ch3).13cnmr(100mhz,cdcl3),δ(ppm):173.24,166.80(c=n),145.29,143.99(ar-c-n),137.32,136.42(ar-cinbackbone),129.58,129.13,128.99,128.93,127.52,127.49(ar-c),125.50,125.36,122.64,121.71(ar-c),50.99,49.50(ch),3.13(pd-ch3).anal.calcdforc29h23cln2pd:c,64.34;h,4.28;n,5.17,其单晶结构示意图如图1。
实施例2
本实施例提供一种α-二亚胺钯配合物2的合成。其反应路线图如图4,具体如下。
二酮的合成与实施例1相同。
配体l2的合成:氮气气氛下,上述步骤制备的二酮(2.34g,10.0mmol,1eq)与2,6-二甲基苯胺(2.7ml,22.0mmol,2.2eq)溶解于100ml甲苯中,加入对甲苯磺酸,加热分水回流24小时。反应后的混合物旋转蒸发除去溶剂,所得固体用乙醇重结晶得到配体l2,产率75%。1hnmr(cdcl3,300mhz),δ(ppm):7.07-7.25(m,12h,ar-h),4.91(s,2h,ch),2.53(m,4h,ch),1.23(s,18h,ch3),1.15(m,12h,ch3),1.03(m,12h,ch3).13cnmr(400mhz,cdcl3),δ(ppm):155.16,143.22,140.34,136.77,128.34,126.65,123.32,122.31,34.50,31.36,30.51,28.94,23.37。
α-二亚胺钯配合物2的合成:α-二亚胺甲基氯化钯配合物2由α-二亚胺配体l2与pd(cod)ch3cl反应得到。具体步骤与实施例1相同,得到的产物为橙红色粉末,反应产率76%。1hnmr(400mhz,cdcl3),δ(ppm):7.30-7.22(m,14h,ar-h),4.95(s,1h,ch),4.90(s,1h,ch),2.79(m,4h,ch),1.47-1.38(d,12h,ch3),1.16-1.12(d,12h,ch3),0.52(s,3h,ch3).13cnmr(400mhz,cdcl3),δ(ppm):171.89,167.03,140.39,140.00,139.35,137.97,137.23,128.66,128.49,128.28,127.55,126.26,126.22,124.10,123.43,51.35,50.48,29.45,29.07,28.66,24.31,23.88,23.39,23.28,3.95。
实施例3
本实施例提供一种α-二亚胺钯配合物3的合成。其反应路线图如图4,具体如下。
二酮的合成与实施例1相同。
配体l3的合成:氮气气氛下,上述步骤制备的二酮(2.34g,10.0mmol,1eq)与2,6-二异丙基苯胺(3.7ml,22.0mmol,2.2eq)溶解于100ml甲苯中,加入对甲苯磺酸,加热分水回流24小时。反应后的混合物旋转蒸发除去溶剂,所得固体用乙醇重结晶得到配体l3,产率82%。1hnmr(400mhz,cdcl3),7.15-7.25(m,14h,ar-h),4.97(d,2h,ch),2.51(m,4h,ch(ch3)2),1.16(d,12h,ch3),1.03(d,12h,ch3).13cnmr(400mhz,cdcl3),δ(ppm):158.44,145.59,136.39,127.30,125.42,124.15,122.81,51.15,28.32,23.29,22.47。
α-二亚胺钯配合物3的合成:α-二亚胺甲基氯化钯配合物3由α-二亚胺配体l3与pd(cod)ch3cl反应得到。具体步骤与实施例1相同,得到的产物为橙色粉末,反应产率73%。1hnmr(400mhz,cdcl3),δ(ppm):7.42-7.22(m,14h,ar-h),4.96(s,1h,ch),4.95(s,1h,ch),2.80(m,4h,ch(ch3)2),1.39(d,6h,ch(ch3)2),1.29(d,6h,ch(ch3)2),1.16-1.12(d,12h,ch(ch3)2),0.52(s,3h,pd-ch3).13cnmr(100mhz,cdcl3),δ(ppm):171.89(c=n),167.03(ar-c-n),140.39(ar-cinbackboneandar-c),140.00(ar-c),139.35(ar-c),137.97,137.23,128.66,128.49,128.28,127.55,126.26,126.22,124.10(ar-cinbackbone),123.43(ar-cinbackbone),51.35(ch),50.48(ch),28.86(ch(ch3)2),28.43(ch(ch3)2),24.31(ch(ch3)2),23.88(ch(ch3)2),23.39(ch(ch3)2),23.28(ch(ch3)2),3.95(pd-ch3).
实施例4
本实施例提供一种α-二亚胺钯配合物4的合成。其反应路线图如图4,具体如下。
二酮的合成与实施例1相同。
配体l4的合成:氮气气氛下,上述步骤制备的二酮(2.34g,10.0mmol,1eq)与4-甲氧基苯胺(2.7ml,22.0mmol,2.2eq)溶解于100ml甲苯中,加入对甲苯磺酸,加热分水回流24小时。反应后的混合物旋转蒸发除去溶剂,所得固体用乙醇重结晶得到配体l4,产率90%。1hnmr(400mhz,cdcl3),δ(ppm):7.36-7.21(m,8h,ar-h),7.03(d,4h,ar-h),6.91(d,4h,ar-h),5.52(s,2h,ch),3.91(s,6h,p-och3).13cnmr(100mhz,cdcl3),δ(ppm):160.91(c=n),156.75(ar-c-p-och3),143.18(ar-c-n),138.67(ar-cinbackbone),127.78(ar-cinbackbone),124.58(ar-cinbackbone),121.51(ar-c),114.42(ar-c),55.19(p-och3),50.01(ch)。
α-二亚胺钯配合物4的合成:α-二亚胺甲基氯化钯配合物4由α-二亚胺配体l4与pd(cod)ch3cl反应得到。具体步骤与实施例1相同,得到的产物为橙红色粉末,反应产率89%。1hnmr(400mhz,cdcl3),δ(ppm):7.40-7.23(m,8h,ar-h),7.18-7.11(d,2h,ar-h),7.11-7.00(m,4h,ar-h),6.94-6.88(d,2h,ar-h),5.47(s,h,ch),5.30(s,ch2cl2),5.08(s,h,ch),3.92(s,3h,p-och3)3.89(s,3h,p-och3),0.68(s,3h,pd-ch3).13cnmr(100mhz,cdcl3),δ(ppm):173.73,166.26(c=n),158.91,158.52(ar-c-p-och3),138.28,137.58(ar-c-n),137.04,136.67,128.95,125.38,124.56,123.07(ar-cinbackbone),114.61,114.08(ar-c),55.50,53.51(p-och3),51.00,49.44(ch),3.32(pd-ch3)。
实施例5
本实施例提供一种α-二亚胺钯配合物5的合成。其反应路线图如图4,具体如下。
二酮的合成与实施例1相同。
配体l5的合成:氮气气氛下,上述步骤制备的二酮(2.34g,10.0mmol,1eq)与3,5-二甲氧基苯胺(3.3ml,22.0mmol,2.2eq)溶解于100,ml甲苯中,加入对甲苯磺酸,加热分水回流24小时。反应后的混合物旋转蒸发除去溶剂,所得固体用乙醇重结晶得到配体l5,产率86%。1hnmr(400mhz,cdcl3),δ(ppm):7.38-7.21(m,8h,ar-h),6.15(s,4h,ar-h),5.50(s,2h,ch),3.93(s,6h,p-och3),3.87(s,12h,m-och3).13cnmr(100mhz,cdcl3),δ(ppm):161.12(c=n),153.68(ar-c-m-och3),146.16(ar-cinbackbone),138.75(ar-c-n),134.55(ar-c-p-och3),127.93,124.86(ar-cinbackbone),97.18(ar-c),61.10(p-och3),56.15(m-och3),50.31(ch)。
α-二亚胺钯配合物5的合成:α-二亚胺甲基氯化钯配合物5由α-二亚胺配体l5与pd(cod)ch3cl反应得到。具体步骤与实施例1相同,得到的产物为橙红色粉末,反应产率86%。1hnmr(400mhz,cdcl3),δ(ppm):7.41-7.27(m,8h,ar-h),6.48(t,1h,ar-h),6.46(t,1h,ar-h),6.30(d,2h,ar-h),6.13(d,2h,ar-h),5.49(s,h,ch),5.17(s,h,ch),3.84(s,12h,m-och3),0.78(s,3h,pd-ch3).13cnmr(100mhz,cdcl3),δ(ppm):173.15,166.87(c=n),161.58,160.86(ar-c-m-och3),146.94,145.68(ar-c-n),137.35,136.49,129.13,128.99,125.51,125.36(ar-cinbackbone),100.95,100.08(ar-c),99.97,99.26(ar-c),55.71,55.65(m-och3),50.99,49.54(ch),3.24(pd-ch3)。
实施例6
本实施例提供一种α-二亚胺钯配合物6的合成。其反应路线图如图4,具体如下。
二酮的合成与实施例1相同。
配体l6的合成:氮气气氛下,上述步骤制备的二酮(2.34g,10.0mmol,1eq)与3,4,5-二甲氧基苯胺(4.0ml,22.0mmol,2.2eq)溶解于100ml甲苯中,加入对甲苯磺酸,加热分水回流24小时。反应后的混合物旋转蒸发除去溶剂,所得固体用乙醇重结晶得到配体l6,产率93%。1hnmr(400mhz,cdcl3),δ(ppm):6.02(s,4h,ar-h),3.85(s,18h,och3),2.20(s,6h,ch3).13cnmr(100mhz,cdcl3),δ(ppm):168.70(c=n),153.04(ar-c-m-och3),147.02(ar-c-p-och3),134.44(ar-c-n),96.03(ar-c),61.02(p-och3),56.08(m-och3),15.61(ch3)。
α-二亚胺钯配合物6的合成:α-二亚胺甲基氯化钯配合物6由α-二亚胺配体l6与pd(cod)ch3cl反应得到。具体步骤与实施例1相同,得到的产物为橙红色粉末,反应产率85%。1hnmr(400mhz,cdcl3),δ(ppm):7.42-7.28(m,8h,ar-h),6.41(s,2h,ar-h),6.21(s,2h,ar-h),5.56(s,h,ch),5.28(s,h,ch),3.97(s,3h,p-och3),3.96(s,3h,p-och3),3.95(s,6h,m-och3),3.90(s,6h,m-och3),0.79(s,3h,pd-ch3).13cnmr(100mhz,cdcl3),δ(ppm):173.55,166.69(c=n),153.94,153.27(ar-c-m-och3),141.08,139.59(ar-c-n),137.49,137.06(ar-c-p-och3),136.60(ar-cinbackbone),129.16,129.05,125.35,125.25(ar-cinbackbone),100.65,99.04(ar-c),61.04(p-och3),56.41(m-och3),53.48(ch2cl2),51.03,49.58(ch),3.42(pd-ch3).anal.calcdforc35h35cln2o6pd:c,58.26;h,4.89;n,3.88。
实施例7
本实施例提供一种超高分子量聚(4-甲氧基苯乙烯),通过如下方法制备得到。
在干燥的玻璃反应瓶中,依次加入1ml4-甲氧基苯乙烯、9ml二氯甲烷,整个体系恒温在30℃;5μmolα-二亚胺钯配合物1和6μmolnabarf在1ml二氯甲烷中混合均匀(单体/催化剂=1490:1),随后加入单体溶液中引发聚合,反应30分钟后加入甲醇/盐酸混合溶液终止反应,过滤,洗涤,干燥,得到聚(4-甲氧基苯乙烯)产物0.80克,单体转化率79.4%。gpc分析测试结果(mw=1063kg/mol,pdi=1.17)。
实施例8
本实施例提供一种超高分子量聚(4-甲氧基苯乙烯),通过如下方法制备得到。
和实施例7相同的聚合条件,5μmolα-二亚胺钯配合物2来代替α-二亚胺钯配合物1。得到聚(4-甲氧基苯乙烯)产物0.89克,单体转化率88.6%。gpc分析测试结果(mw=1024kg/mol,pdi=1.20)。
实施例9
本实施例提供一种超高分子量聚(4-甲氧基苯乙烯),通过如下方法制备得到。
和实施例7相同的聚合条件,5μmolα-二亚胺钯配合物3来代替α-二亚胺钯配合物1。得到聚(4-甲氧基苯乙烯)产物0.99克,单体转化率98.1%。gpc分析测试结果(mw=1022kg/mol,pdi=1.15),其核磁氢谱图和核磁碳谱图分别如图2和图3所示。
实施例10
本实施例提供一种超高分子量聚(4-甲氧基苯乙烯),通过如下方法制备得到。
和实施例7相同的聚合条件,5μmolα-二亚胺钯配合物4来代替α-二亚胺钯配合物1。得到聚(4-甲氧基苯乙烯)产物0.80克,单体转化率80.0%。gpc分析测试结果(mw=1141kg/mol,pdi=1.22)。
实施例11
本实施例提供一种超高分子量聚(4-甲氧基苯乙烯),通过如下方法制备得到。
和实施例7相同的聚合条件,5μmolα-二亚胺钯配合物5来代替α-二亚胺钯配合物1。得到聚(4-甲氧基苯乙烯)产物0.78克,单体转化率77.3%。gpc分析测试结果(mw=1064kg/mol,pdi=1.24)。
实施例12
本实施例提供一种超高分子量聚(4-甲氧基苯乙烯),通过如下方法制备得到。
和实施例7相同的聚合条件,5μmolα-二亚胺钯配合物6来代替α-二亚胺钯配合物1。得到聚(4-甲氧基苯乙烯)产物0.79克,单体转化率79.2%。gpc分析测试结果(mw=964kg/mol,pdi=1.17)。
实施例13
本实施例提供一种超高分子量聚(4-乙氧基苯乙烯),通过如下方法制备得到。
和实施例7相同的聚合条件,1ml4-乙氧基苯乙烯单体来代替4-甲氧基苯乙烯。得到聚(4-乙氧基苯乙烯)产物0.80克,单体转化率80.2%。gpc分析测试结果(mw=1205kg/mol,pdi=1.20)。
实施例14
本实施例提供一种超高分子量聚(4-乙氧基苯乙烯),通过如下方法制备得到。
和实施例8相同的聚合条件,1ml4-乙氧基苯乙烯单体来代替4-甲氧基苯乙烯。得到聚(4-乙氧基苯乙烯)产物0.88克,单体转化率88.1%。gpc分析测试结果(mw=1315kg/mol,pdi=1.19)。
实施例15
本实施例提供一种超高分子量聚(4-乙氧基苯乙烯),通过如下方法制备得到。
和实施例9相同的聚合条件,1ml4-乙氧基苯乙烯单体来代替4-甲氧基苯乙烯。得到聚(4-乙氧基苯乙烯)产物0.98克,单体转化率98.2%。gpc分析测试结果(mw=1005kg/mol,pdi=1.17),其核磁氢谱图和核磁碳谱图分别如图2和图3所示。
实施例16
本实施例提供一种超高分子量聚(4-乙氧基苯乙烯),通过如下方法制备得到。
和实施例10相同的聚合条件,1ml4-乙氧基苯乙烯单体来代替4-甲氧基苯乙烯。得到聚(4-乙氧基苯乙烯)产物0.78克,单体转化率78.1%。gpc分析测试结果(mw=1213kg/mol,pdi=1.22)。
实施例17
本实施例提供一种超高分子量聚(4-乙氧基苯乙烯),通过如下方法制备得到。
和实施例11相同的聚合条件,1ml4-乙氧基苯乙烯单体来代替4-甲氧基苯乙烯。得到聚(4-乙氧基苯乙烯)产物0.78克,单体转化率78.1%。gpc分析测试结果(mw=1213kg/mol,pdi=1.22)。
实施例18
本实施例提供一种超高分子量聚(4-乙氧基苯乙烯),通过如下方法制备得到。
和实施例12相同的聚合条件,1ml4-乙氧基苯乙烯单体来代替4-甲氧基苯乙烯。得到聚(4-乙氧基苯乙烯)产物0.81克,单体转化率81.1%。gpc分析测试结果(mw=1200kg/mol,pdi=1.21)。
实施例19
本实施例提供一种超高分子量聚(4-甲氧基苯乙烯),通过如下方法制备得到。
和实施例9相同的聚合条件,活化剂用6μmolagpf6来代替nabarf。得到聚(4-甲氧基苯乙烯)产物0.91克,单体转化率90.2%。gpc分析测试结果(mw=1003kg/mol,pdi=1.25)。
实施例20
本实施例提供一种超高分子量聚(4-甲氧基苯乙烯),通过如下方法制备得到。
和实施例9相同的聚合条件,活化剂用6μmolagsbf6来代替nabarf。得到聚(4-甲氧基苯乙烯)产物0.95克,单体转化率94.2%。gpc分析测试结果(mw=1039kg/mol,pdi=1.22)。
实施例21
本实施例提供一种超高分子量聚(4-甲氧基苯乙烯),通过如下方法制备得到。
和实施例9相同的聚合条件,用5μmolnabarf来代替6μmolnabarf。得到聚(4-甲氧基苯乙烯)产物0.94克,单体转化率93.2%。gpc分析测试结果(mw=1022kg/mol,pdi=1.19)。
实施例22
本实施例提供一种超高分子量聚(4-甲氧基苯乙烯),通过如下方法制备得到。
和实施例9相同的聚合条件,用10μmolnabarf来代替6μmolnabarf。得到聚(4-甲氧基苯乙烯)产物0.98克,单体转化率97.1%。gpc分析测试结果(mw=1112kg/mol,pdi=1.24)。
实施例23
本实施例提供一种超高分子量聚(4-甲氧基苯乙烯),通过如下方法制备得到。
和实施例9相同的聚合条件,用20μmolnabarf来代替6μmolnabarf。得到聚(4-甲氧基苯乙烯)产物0.97克,单体转化率96.1%。gpc分析测试结果(mw=1032kg/mol,pdi=1.21)。
实施例24
本实施例提供一种超高分子量聚(4-甲氧基苯乙烯),通过如下方法制备得到。
和实施例9相同的聚合条件,用25μmolnabarf来代替6μmolnabarf。得到聚(4-甲氧基苯乙烯)产物0.93克,单体转化率92.1%。gpc分析测试结果(mw=1169kg/mol,pdi=1.20)。
实施例25
本实施例提供一种超高分子量聚(4-甲氧基苯乙烯),通过如下方法制备得到。
和实施例9相同的聚合条件,溶剂用1,1,2,2-四氯乙烷来代替二氯甲烷。得到聚(4-甲氧基苯乙烯)产物0.98克,单体转化率97.1%。gpc分析测试结果(mw=1123kg/mol,pdi=1.20)。
实施例26
本实施例提供一种超高分子量聚(4-甲氧基苯乙烯),通过如下方法制备得到。
和实施例9相同的聚合条件,溶剂用氯苯来代替二氯甲烷。得到聚(4-甲氧基苯乙烯)产物0.88克,单体转化率87.2%。gpc分析测试结果(mw=1029kg/mol,pdi=1.22)。
实施例27
本实施例提供一种超高分子量聚(4-甲氧基苯乙烯),通过如下方法制备得到。
和实施例9相同的聚合条件,溶剂用甲苯来代替二氯甲烷。得到聚(4-甲氧基苯乙烯)产物0.91克,单体转化率90.2%。gpc分析测试结果(mw=1069kg/mol,pdi=1.19)。
实施例28
本实施例提供一种超高分子量聚(4-甲氧基苯乙烯),通过如下方法制备得到。
和实施例9相同的聚合条件,单体用0.5ml4-甲氧基苯乙烯来代替1ml4-甲氧基苯乙烯(单体/催化剂=750:1)。得到聚(4-甲氧基苯乙烯)产物0.35克,单体转化率69.3%。gpc分析测试结果(mw=1001kg/mol,pdi=1.26)。
实施例29
本实施例提供一种超高分子量聚(4-甲氧基苯乙烯),通过如下方法制备得到。
和实施例9相同的聚合条件,单体用2ml4-甲氧基苯乙烯来代替1ml4-甲氧基苯乙烯(单体/催化剂=2980:1)。得到聚(4-甲氧基苯乙烯)产物1.87克,单体转化率92.7%。gpc分析测试结果(mw=1560kg/mol,pdi=1.26)。
实施例30
本实施例提供一种超高分子量聚(4-甲氧基苯乙烯),通过如下方法制备得到。
和实施例9相同的聚合条件,单体用4ml4-甲氧基苯乙烯来代替1ml4-甲氧基苯乙烯(单体/催化剂=5960:1)。得到聚(4-甲氧基苯乙烯)产物3.57克,单体转化率88.4%。gpc分析测试结果(mw=1960kg/mol,pdi=1.29)。
实施例31
本实施例提供一种超高分子量聚(4-甲氧基苯乙烯),通过如下方法制备得到。
和实施例9相同的聚合条件,单体用6.7ml4-甲氧基苯乙烯来代替1ml4-甲氧基苯乙烯(单体/催化剂=10000:1)。得到聚(4-甲氧基苯乙烯)产物6.35克,单体转化率93.9%。gpc分析测试结果(mw=4598kg/mol,pdi=1.30)。
实施例32
本实施例提供一种超高分子量聚(4-甲氧基苯乙烯),通过如下方法制备得到。
和实施例23相同的聚合条件,反应温度用0℃来代替30℃。得到聚(4-甲氧基苯乙烯)产物0.45克,单体转化率44.6%。gpc分析测试结果(mw=1012kg/mol,pdi=1.12)。
实施例33
本实施例提供一种超高分子量聚(4-甲氧基苯乙烯),通过如下方法制备得到。
和实施例23相同的聚合条件,反应温度用50℃来代替30℃。得到聚(4-甲氧基苯乙烯)产物0.97克,单体转化率96.1%。gpc分析测试结果(mw=1236kg/mol,pdi=1.26)。
实施例34
本实施例提供一种超高分子量聚(4-甲氧基苯乙烯),通过如下方法制备得到。
和实施例23相同的聚合条件,反应温度用80℃来代替30℃。得到聚(4-甲氧基苯乙烯)产物0.97克,单体转化率96.1%。gpc分析测试结果(mw=1236kg/mol,pdi=1.26)。
对比例1
本对比例与实施例9相同的聚合条件,用经典骨架的α-二亚胺钯催化剂(结构式如下式iii所示)代替α-二亚胺钯催化剂3,得到聚(4-甲氧基苯乙烯)0.014克,单体转化率1.3%。gpc分析测试结果(mw=25kg/mol,pdi=2.19)。
由此证明了通过在骨架上引入大位阻刚性的二苯并桶烯骨架在聚合反应中起着关键的作用,能够显著的提高聚合活性,单体的转化率相对于对比例提高了近90倍,同时制备得到的聚合物分子量相对于对比例,分子量大大提高,并且分子量分布更窄。
以实施例9制备的聚合物(超高分子量聚(4-甲氧基苯乙烯))为例,对其性能进行测定及与商用聚苯乙烯进行对比,如表1所示:
表1聚(4-甲氧基苯乙烯)的性能测定结果
a聚合物降解5%时的温度。
b聚合物降解10%时的温度。
由表1中的数据可以得出,超高分子量的聚(4-烷氧基苯乙烯)与对比商用聚苯乙烯(中石化sp-666d)相比,拉伸强度和断裂伸长率分别提高了26.1%和41.5%;5%的聚合物降解温度从360.9℃提升到392.1℃,玻璃化转变温度也从94.7提升到110.8℃,聚合物的热稳定性能明显提高;接触角从98.6降低到61.6°,亲水性能大大增强。综合考虑,本发明制备的聚(4-烷氧基苯乙烯)与商用聚苯乙烯相比具有更好的机械性能、热稳定性能和亲水性能。
上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的限制,其他的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!