一种唑吡坦杂质的制备方法与流程
本发明属于化学合成
技术领域:
。更具体地,涉及一种唑吡坦杂质的制备方法。
背景技术:
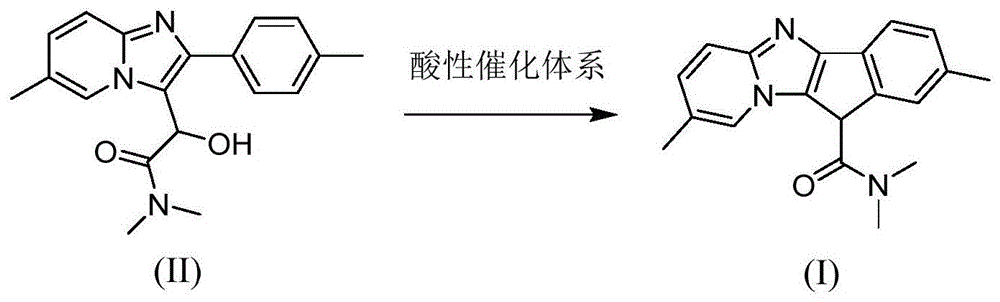
:唑吡坦是新一代非苯二氮卓类的短效咪唑并吡啶类的镇静催眠药,临床上以酒石酸唑吡坦形式口服给药,用于治疗失眠以及脑部疾病。作用原理是可以选择性地与中枢神经系统的ω1-受体亚型结合,使氯离子通道开放,让氯离子流入神经细胞内,引起细胞膜超级化,从而抑制神经细胞源激动,与苯二氮卓类药物相比,具有更高的选择性。并且长期临床试验表明,唑吡坦在正常的治疗周期内极少产生耐受性和成瘾性,也极少有记忆障碍、抑郁综合征、精神障碍等不良反应,与具有多种不良反应和戒断症状的苯二氮卓类药物相比,具有更高的安全性,应用前景更加广阔。目前,唑吡坦的主流合成路线为:在上述合成过程中,化合物ii中的羟基在氯化亚砜存在条件下发生卤代反应,形成的酸性物质发生傅克烷基化反应,最终生成杂质化合物ⅰ。生成的杂质化合物i存在于终产品中,影响唑吡坦原料药的纯度,需要在合成唑吡坦过程中对该杂质进行检测监控,从而控制唑吡坦原料药的质量。但是杂质化合物i在唑吡坦的合成过程中含量非常低,难以分离,且现有技术尚无针对杂质化合物i的合成方法,难以获得大量的化合物i,缺少相应的对照品,在唑吡坦合成过程中难以对该杂质的检测进行监控。因此,迫切需要提供一种唑吡坦杂质化合物i的制备方法。技术实现要素:本发明要解决的技术问题是克服现有技术缺少唑吡坦杂质化合物i合成方法,缺少杂质化合物i对照品的缺陷和不足,提供一种唑吡坦杂质化合物i的制备方法。本发明上述目的通过以下技术方案实现:一种唑吡坦杂质的制备方法,反应路线如下:具体包括以下步骤:在惰性气体保护下,将化合物ii与酸性催化体系混合,于80~140℃反应,后处理、纯化,得化合物i;其中,所述酸性催化体系为多聚磷酸体系、或98%浓硫酸和非质子类有机溶剂体系、或三氯化铝和非质子类有机溶剂体系。本发明中,采用多聚磷酸体系、或98%浓硫酸和非质子类有机溶剂体系、或三氯化铝和非质子类有机溶剂体系作为反应的酸性催化体系,使化合物ii发生傅克烷基化反应得到化合物i。具体反应原理为:在特定的酸性催化条件下,化合物ⅱ首先形成稳定的碳正离子,然后进攻富电子上的苯环,取代苯环上的氢,从而成环形成化合物i。进一步地,当酸性催化体系为多聚磷酸体系时,所述化合物ii与多聚磷酸的质量体积比为1:(5~30)g/ml。实践中发现,当酸性催化体系为多聚磷酸体系时,反应速率和收率较高,其原因可能是多聚磷酸具有一定的脱水作用,可以促进反应向生成化合物i的方向进行。优选地,当酸性催化体系为多聚磷酸体系时,所述化合物ii与多聚磷酸的质量体积比为1:(5~10)g/ml。在实践中发现,在此质量体积比范围中制备得到的化合物i收率较高。更优选地,当酸性催化体系为多聚磷酸体系时,所述化合物ii与多聚磷酸的质量体积比为1:5g/ml。在实践中发现,在此条件下制备得到的化合物i收率最高。优选地,当酸性催化体系为多聚磷酸体系时,所述反应的温度为100~120℃。实践中发现,在此温度范围中制备得到的化合物i收率较高。更进一步地,当酸性催化体系为多聚磷酸体系时,所述反应的时间为2~6h。进一步地,所述非质子类有机溶剂选自二氯乙烷、甲苯、二甲基甲酰胺中的一种。更进一步地,当酸性催化体系为98%浓硫酸和非质子类有机溶剂体系时,所述反应的温度为80~85℃。实践中发现,在此温度范围中制备得到的化合物i收率较高。优选地,当酸性催化体系为98%浓硫酸和非质子类有机溶剂体系时,所述非质子类有机溶剂为二氯乙烷。实践中发现,98%浓硫酸和二氯乙烷作为酸性催化体系制备得到的化合物i收率较高。进一步地,当酸性催化体系为98%浓硫酸和非质子类有机溶剂体系时,所述化合物ii与98%浓硫酸的物质的量比为1:(3~6)。优选地,当酸性催化体系为98%浓硫酸和非质子类有机溶剂体系时,所述化合物ii与98%浓硫酸的物质的量比为1:(4~6)。更优选地,当酸性催化体系为98%浓硫酸和非质子类有机溶剂体系时,所述化合物ii与98%浓硫酸的物质的量比为1:5。实践中发现,在此条件下,化合物ii与酸性催化体系可充分接触,提高反应效率。更进一步地,当酸性催化体系为98%浓硫酸和非质子类有机溶剂体系时,所述化合物ii和非质子类有机溶剂的质量体积比为1:(2~6)。进一步地,当酸性催化体系为98%浓硫酸和非质子类有机溶剂体系时,所述反应的时间为5~20h。优选地,当酸性催化体系为三氯化铝和非质子类有机溶剂体系时,所述反应的温度为100~120℃。实践中发现,在此温度范围中制备得到的化合物i收率较高。优选地,当酸性催化体系为三氯化铝和非质子类有机溶剂体系时,所述非质子类有机溶剂为二甲基甲酰胺。实践中发现,三氯化铝和二甲基甲酰胺作为酸性催化体系制备得到的化合物i收率较高。更进一步地,当酸性催化体系为三氯化铝和非质子类有机溶剂体系时,所述化合物ii与三氯化铝的物质的量比为1:(3~6)。优选地,当酸性催化体系为三氯化铝和非质子类有机溶剂体系时,所述化合物ii与三氯化铝的物质的量比为1:(4~6)。更优选地,当酸性催化体系为三氯化铝和非质子类有机溶剂体系时,所述化合物ii与三氯化铝的物质的量比为1:5。实践中发现,在此条件下,化合物ii与酸性催化体系可充分接触,提高反应效率。进一步地,当酸性催化体系为三氯化铝和非质子类有机溶剂体系时,所述化合物ii和非质子类有机溶剂的质量体积比为1:(2~6)。更进一步地,当酸性催化体系为三氯化铝和非质子类有机溶剂体系时,所述反应的时间为12~60h。本发明具有以下有益效果:本发明提供了一种全新的唑吡坦杂质的制备方法,采用化合物ii(n,n,6-三甲基-2-(4-甲基苯基)-咪唑并[1,2-α]吡啶-3-α-羟基乙酰胺)作为原料,结合特定的多聚磷酸、浓硫酸、三氯化铝酸性催化体系,在80~140℃条件下进行反应,产物收率较高,且操作简单,适合大规模产业化生产唑吡坦杂质化合物i,有利于合成唑吡坦过程中对该杂质的检测监控。附图说明图1为本发明唑吡坦杂质化合物i制备方法的合成路线图。具体实施方式以下结合说明书附图和具体实施例来进一步说明本发明,但实施例并不对本发明做任何形式的限定。除非特别说明,本发明采用的试剂、方法和设备为本
技术领域:
常规试剂、方法和设备。除非特别说明,以下实施例所用试剂和材料均为市购。本发明唑吡坦杂质化合物i制备方法的合成路线图如下所示:其中,化合物ii由以下合成路线制备:具体包括以下步骤:将4.00gn,n-二甲基-2,2-二甲氧基乙酰胺、1.22ml水、5.57g醋酸钠、5.67ml浓盐酸(质量分数为37%)投入反应瓶中,搅拌,升温至43~46℃活化50min后,加入3.02g6-甲基-2-(4-甲基苯基)-咪唑并[1,2-α]吡啶、15ml1,2-二氯乙烷,搅拌,升温至76~80℃,回流3h;反应结束后降温至60℃以下,用饱和na2co3溶液调节ph=7,减压浓缩,加20ml异丙醇水溶液(异丙醇:水=1:1),升温至80~90℃,回流20min,静置30min,分液,上层液体冷却析晶5h,过滤,滤渣用水洗涤、干燥,得3.10g白色固体物化合物ii(n,n,6-三甲基-2-(4-甲基苯基)-咪唑并[1,2-α]吡啶-3-α-羟基乙酰胺),收率为70.54%。实施例1一种唑吡坦杂质化合物i的制备采用如下方法制备分离得到所述唑吡坦杂质化合物i:在n2保护下,将970mg化合物ii(1.0eq,3mmol)与5ml多聚磷酸搅拌混合,于100℃反应2h,反应结束后,加入5ml冰水,用10ml乙酸乙酯分两次萃取,无水硫酸钠干燥,减压浓缩,用硅胶拌样,进行柱层析(展开剂为乙酸乙酯:石油醚=1:3),得742mg类白色固体,esi:m/z[m+h]+306.10,1hnmr(cdcl3):δ2.35(s,3h),2.56(s,3h),3.27(s,3h),3.47(s,3h),6.36(s,1h),6.86-6.91(m,2h),7.16-7.19(m,1h),7.44-7.49(m,2h),8.06-80.8(m,1h),为唑吡坦杂质化合物i,收率为81%。参考上述制备方法,不同之处在于,采用不同添加量多聚磷酸或反应温度制备唑吡坦杂质化合物i,计算所得唑吡坦杂质化合物i的收率。具体多聚磷酸添加量、反应温度和实验结果参见表1。表1不同添加量多聚磷酸或反应温度对唑吡坦杂质化合物i收率的影响序号化合物ii:多聚磷酸(g/ml)反应温度(℃)收率(%)1(实施例1)1:51108121:81107831:101107741:151106851:201105561:251104671:301103081:5805691:510080101:512081111:514074由表1可见,当酸性催化体系为多聚磷酸体系时,在相同的温度条件下,随着多聚磷酸的添加量增加,杂质化合物i的收率也随着显著降低;在相同多聚磷酸添加量的条件下,反应温度在100~120℃时,收率较高。实施例2一种唑吡坦杂质化合物i的制备采用如下方法制备分离得到所述唑吡坦杂质化合物i:在n2保护下,将970mg化合物ii(1.0eq,3mmol)与5ml二氯乙烷搅拌混合,加入815μl98%浓硫酸(5.0eq,15mmol),于80℃反应5h,反应结束后,加入5ml冰水,用10ml二氯甲烷分两次萃取,无水硫酸钠干燥,减压浓缩,用硅胶拌样,进行柱层析(展开剂为乙酸乙酯:石油醚=1:3),得623mg类白色固体,esi:m/z[m+h]+306.10,1hnmr(cdcl3):δ2.35(s,3h),2.56(s,3h),3.27(s,3h),3.47(s,3h),6.36(s,1h),6.86-6.91(m,2h),7.16-7.19(m,1h),7.44-7.49(m,2h),8.06-80.8(m,1h),为唑吡坦杂质化合物i,收率为68%。参考上述制备方法,不同之处在于,采用不同非质子类有机溶剂或反应温度制备唑吡坦杂质化合物i,计算所得唑吡坦杂质化合物i的收率。具体非质子类有机溶剂、反应温度和实验结果参见表2。表2不同非质子类有机溶剂或反应温度对唑吡坦杂质化合物i收率的影响序号非质子类有机溶剂反应温度(℃)收率(%)1(实施例2)二氯乙烷80682二氯乙烷85677甲苯80508二甲基甲酰胺8045由表2可见,当酸性催化体系为98%浓硫酸和非质子类有机溶剂体系时,在相同的温度条件下,以二氯乙烷制备的杂质化合物i的收率较高;在相同非质子类有机溶剂的条件下,反应温度在80~85℃时,收率较高。实施例3一种唑吡坦杂质化合物i的制备采用如下方法制备分离得到所述唑吡坦杂质化合物i:在n2保护下,将970mg化合物ii(1.0eq,3mmol)与5ml二甲基甲酰胺搅拌混合,加入2.0g三氯化铝(5.0eq,15mmol),于100℃反应12h,反应结束后,浓缩后加入乙酸乙酯,重复浓缩2~3次,用硅胶拌样,进行柱层析(展开剂为乙酸乙酯:石油醚=1:3),得567mg类白色固体,esi:m/z[m+h]+306.10,1hnmr(cdcl3):δ2.35(s,3h),2.56(s,3h),3.27(s,3h),3.47(s,3h),6.36(s,1h),6.86-6.91(m,2h),7.16-7.19(m,1h),7.44-7.49(m,2h),8.06-80.8(m,1h),为唑吡坦杂质化合物i,收率为62%。参考上述制备方法,不同之处在于,采用不同非质子类有机溶剂或反应温度制备唑吡坦杂质化合物i,计算所得唑吡坦杂质化合物i的收率。具体非质子类有机溶剂、反应温度和实验结果参见表3。表3不同非质子类有机溶剂或反应温度对唑吡坦杂质化合物i收率的影响序号非质子类有机溶剂反应温度(℃)收率(%)1(实施例3)二甲基甲酰胺100622甲苯100553二氯乙烷80454二甲基甲酰胺110615二甲基甲酰胺120606二甲基甲酰胺14055由表3可见,当酸性催化体系为三氯化铝和非质子类有机溶剂体系时,在相同的温度条件下,以二甲基甲酰胺制备的杂质化合物i的收率较高;在相同非质子类有机溶剂的条件下,反应温度在100~120℃时,收率较高。上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的限制,其他的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1