1-氯-1-氯乙酰基环丙烷的制备方法与流程
本发明涉及精细有机合成领域,具体而言,本发明涉及一种丙硫菌唑中间体1-氯-1-氯乙酰基环丙烷的制备方法。
背景技术:
丙硫菌唑是一种广泛应用的广谱三唑硫酮类杀菌剂,可用于谷类、麦类、豆类等作物的病害,具有高效、低毒、无三致(致癌、致畸、致突变),对人畜影响小,环境安全的优点。目前,国内外多家企业正在或者准备生产该农药品种。
1-氯-1-氯乙酰基环丙烷(casno:120983-72-4)是丙硫菌唑生产工艺中的关键中间体,生产技术主要是以乙酰基-γ-丁内酯为起始原料,经过氯代、酸解、关环、二次氯化等四步得到产品。如美国专利us4938791、中国发明专利cn105384617、cn108586220等。由于生产过程需要连续用到氯气/磺酰氯、烧碱、浓盐酸等,产生大量的三废,对环境破坏较大。目前已有多家高校、企业、科研院所提出了相关的改进方案,如中国发明专利cn105384617、中国发明专利cn107118090等。
考虑到以上丙硫菌唑中间体制备中面临的问题,仍需要开发一种可以以环保方式合成丙硫菌唑中间体的新工艺。
技术实现要素:
本发明的目的是提供一种丙硫菌唑中间体1-氯-1-氯乙酰基环丙烷的制备新工艺,其可以减少强酸、强碱和氯化试剂的使用,进而减少三废排放和降低安全风险,同时可以提高合成收率,以适应工业化生产。
本发明提供一种1-氯-1-氯乙酰基环丙烷的制备方法,其包括以下步骤:
(1)环烷基化反应:使1,3-丁二烯和重氮甲烷反应,生成乙烯基环丙烷;
(2)氧化反应:使乙烯基环丙烷与氧化试剂反应生成乙酰基环丙烷;以及
(3)氯化反应:使乙酰基环丙烷与氯化试剂反应生成目标产物1-氯-1-氯乙酰基环丙烷。
在具体实施方式中,在步骤(1)中,使用的催化剂可选自二氯化钯、醋酸钯或三(二亚苄基茚丙酮)二钯,优选为醋酸钯,并且所述催化剂的用量占1,3-丁二烯重量的1~20%。
在具体实施方式中,在步骤(1)中,使用的溶剂可选自乙醚、二氯甲烷、乙腈或氯仿,优选为乙醚或乙腈。
在具体实施方式中,在步骤(2)中,所述氧化试剂可选自氧气、空气或双氧水,优选为氧气。
在具体实施方式中,在步骤(2中,使用的催化剂可选自氯化钯、醋酸钯,助催化剂可选自氯化亚铜、醋酸亚铜或硝酸银,优选地,使用氯化钯和氯化亚铜作为催化体系,并且催化剂体系用量为乙烯基环丙烷重量的1~10%。
在具体实施方式中,在步骤(2)中使用的溶剂为水-有机溶剂混合体系,在该体系中,所述有机溶剂可选自n,n-二甲基甲酰胺(dmf)、n,n-二甲基乙酰胺、二甲基亚砜(dmso)、乙腈、四氢呋喃或1,4-二氧六环,优选地,所述有机溶剂为dmf;在所述水-有机溶剂混合体系中,水和有机溶剂的体积比为1:10至10:1,优选为1:1,例如所述水-有机溶剂混合体系为水和dmf体积比为1:1的溶剂体系。
在具体实施方式中,在步骤(3)中,所述氯化试剂可选自氯气、磺酰氯、二氯亚砜、烷基磺酰氯或n-氯代丁二酰亚胺(ncs),优选为ncs。
在具体实施方式中,在步骤(3)中使用的溶剂可选自二氯甲烷、氯仿、四氯化碳、乙腈或n,n-二甲基甲酰胺(dmf),优选为二氯甲烷。
在具体实施方式中,步骤(1)的反应温度为0~40℃,优选为10~30℃;
步骤(2)的反应温度为0~80℃,优选为25~45℃,更优选为40℃;以及
步骤(3)的反应温度为0~80℃,优选为20~30℃,更优选为25℃。
在具体实施方式中,在步骤(3)中,氯化试剂的加入方式及加入量如下:在0℃下相对于1摩尔的乙酰基环丙烷,加入1.0~1.1摩尔的氯化试剂,当温度升至室温后,再次加入相对于1摩尔的乙酰基环丙烷1.0~1.1摩尔的氯化试剂。
有益效果
本发明的丙硫菌唑中间体的制备方法具有以下优点:
1)以石化工业常见产品1,3-丁二烯代替了乙酰基-γ-丁内酯作为原料,原料来源更加广泛,易得,节约原料成本;
2)环烷基化试剂使用重氮甲烷,副产物仅为氮气,简化了废气的处理,催化剂和溶剂可以重复利用;
3)乙酰基环丙烷的制备使用空气等作为氧化试剂,原子利用率高,三废排放少,催化剂可多次循环使用;
4)氯化步骤优选使用ncs作为氯化试剂,具有较高的选择性,副产物为琥珀酰亚胺,经简单蒸馏提纯后,可联合氯碱企业加以再生,循环使用;
5)相比较传统工艺,本方法基本没有废水产生,因此,整个工艺绿色环保。
附图说明
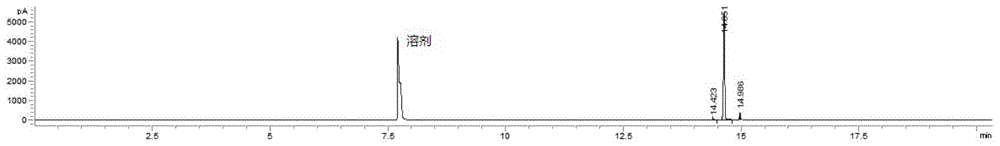
图1为本发明中得到的产物1-氯-1-氯乙酰基环丙烷的气相色谱图。
图2为本发明中得到的产物1-氯-1-氯乙酰基环丙烷的核磁共振图谱。
具体实施方式
下面结合具体实施案例对本发明公开的工艺做出说明。但这些说明不意图限制本发明的范围。
实施例
步骤1:在一个带有四氟内衬的不锈钢反应釜中,加入重氮甲烷的乙醚溶液和催化量的醋酸钯,随后封闭反应釜,并用氮气置换,开启搅拌,在室温下慢慢通入1,3-丁二烯,保持压力为0.4mpa。随着反应进行,压力开始下降,继续补充1,3-丁二烯,直至压力维持0.4mpa不下降后,继续反应1小时。反应结束后,打开反应釜,蒸馏反应液,蒸馏出乙醚和产品乙烯基环丙烷,催化剂醋酸钯留在釜底,可随同蒸馏出的乙醚一同回用。常压下,产品乙烯基环丙烷的沸程为40–42℃。以重氮甲烷消耗计算,该反应收率>90%。
1h-nmr(400mh,cdcl3):δ=5.44(1h,t),5.16(1h,m),4.94(1h,m),1.40(1h,m),0.72(2h,m),0.48(2h,m)。
步骤2:在油浴中,放置一个500ml的三口烧瓶中,安装气体通入-接收装置,并加入四氟搅拌子,随后烧瓶中放入200ml的水和dmf的混合溶剂(体积比1:1),加入20g(0.294mol)的乙烯基环丙烷(步骤1的产品),加入0.5g氯化钯和0.5g的氯化亚铜,开启加热装置,体系升温至40℃,开始通入氧气,氧气流速为20ml/min。反应时间为6小时,期间,用气相色谱检测反应进行的程度。待反应物完全转化后,将装置改为分馏装置,采集不同阶段的样品,可收集得到浅黄色油状液体19g,收率为80%,含量>98%。
1h-nmr(400mh,cdcl3):δ=2.20(3h,s),2.00(1h,m),0.92(2h,m),0.64(2h,m)。
步骤3:在250ml三口圆底烧瓶中加入10g乙酰基环丙烷(0.12mol)和30ml二氯甲烷,置入0℃冷浴下降温,随后加入33.5g的ncs(0.25mol),开启搅拌,慢慢升至室温。搅拌2-3小时。反应生成大量沉淀。反应结束后,在通风橱内过滤。滤饼用少量的二氯甲烷洗涤,合并滤液,滤液蒸馏除去溶剂二氯甲烷,随后减压蒸馏得到目标产物,为浅黄色透明油状液体16.8g,经gc-ms(图1)和h-nmr(图2)确认为产物1-氯-1-氯乙酰基环丙烷,纯度>97%。收率为92%。回收的琥珀酰亚胺可直接用于制备ncs,回用。
1h-nmr(400mh,cdcl3):δ=4.76(2h,s),1.76(2h,q),1.45(2h,q)。
由上述实施例可以看出,本发明的工艺流程简单,综合收率高,可以用于制备高纯度的1-氯-1-氯乙酰基环丙烷,适合工业规模的生产。
以上仅为本发明的具体实施例,任何熟悉本领域的技术人员应用本发明的方法所做出显而易见的变化或改变方式,均在本专利的保护范围内。
- 还没有人留言评论。精彩留言会获得点赞!