基于引入额外碱基错配的阻滞物进行阻滞取代扩增富集检测目标突变的方法与流程
本发明涉及基因测序领域,具体涉及一种对目标区域基因突变进行富集和检测的方法,尤其涉及一种基于引入额外碱基错配的阻滞物进行阻滞取代扩增富集检测目标突变的方法。
背景技术:
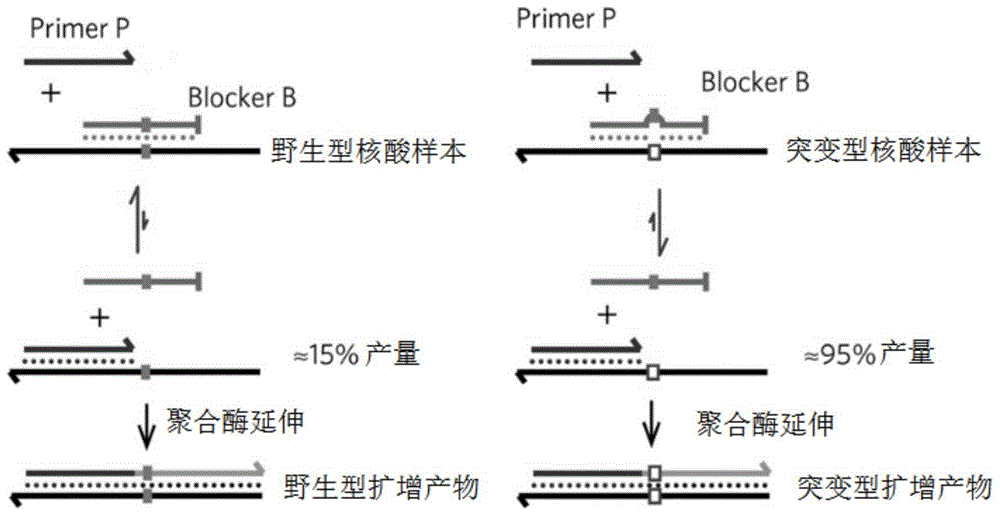
:随着时代和科技的快速发展,癌症治疗观念正在发生根本性的改变。国际抗癌联盟认为,1/3的癌症是可以预防的,1/3的癌症如能早期诊断是可以治愈的,1/3的癌症可以通过正确治疗减轻痛苦,延长生命。癌症的治疗手段也由经验科学向循证医学、由细胞攻击模式向靶向性治疗模式转变。早期传统的治疗方式为手术切除、化疗和放疗,其中化疗和放疗不能区分肿瘤细胞和正常细胞,对人体伤害较大。肿瘤靶向治疗技术能够在无创或微创条件下以肿瘤为目标,采用有选择、针对性较强、患者易于接受、反应小的局部或全身治疗,最终达到有效控制肿瘤的目的,对人体伤害较小。现代的癌症精准医疗就是在患者确诊癌症后进行基因检测来确定基因突变种类,寻找适用于该种突变的靶向治疗药物,最后通过结合患者临床表现来制定正确的靶向治疗方案来治疗癌症。基因检测作为靶向治疗的上游事件,对指导整个靶向治疗方案起到十分重要的作用。起初,我们使用肿瘤组织作为基因检测的材料,但是因为获取肿瘤组织对人体伤害较大,部分患者的状况不适宜获取肿瘤组织等原因,我们使用血液作为替代的基因检测材料。1948年,人们首次发现人体血液中存在游离dna(cfdna),这是一种以单链或双链dna或dna蛋白复合物形式存在于血液、脑脊液中的一种胞外dna。在肿瘤患者体内,由肿瘤细胞坏死、凋亡后释放入血液中的游离dna片段称为循环肿瘤dna(ctdna)。ctdna片段大小一般为160-180bp左右,半衰期为16min-2h不等。由于ctdna携带了肿瘤细胞的全部变异信息,且这些基因突变仅存在于癌前细胞或癌细胞基因组中,不会在同一机体的其他正常细胞dna中出现,因此,ctdna相对于传统的组织学样本能够更好的体现肿瘤异质性。此外,ctdna无需侵入性取材,只需取一管血即可检测,能够方便地监测肿瘤进程、监测靶向治疗动态。因此,基于ctdna的肿瘤基因检测技术逐渐成为临床应用的热点,具有广泛前景。目前,市面上的许多的ctdna检测技术,包括ngs检测技术、基于arms原理的qpcr检测技术以及数字pcr等。其中ngs检测最为广泛。ctdna检测除了在指导靶向治疗上起到重要作用,在癌症的术后监控以及癌症的早期筛查方便也具有巨大的潜力,但要将其广泛应用于临床还需克服诸多障碍。首先,血液中游离dna含量很少,平均1ml血浆中只能提取到约10ng左右总游离dna,ctdna占总游离dna只有0.1%-10%,而在癌症术后病人和早期癌症病人的血液中ctdna占比还要更低的多。所以,目前急需一种既能完成低频突变位点检测又能达到一定通量的ctdna检测技术,市面上主要存在的这几种ctdna检测技术均不能满足要求。这种一定通量的低频突变位点检测是目前ctdna检测技术遇到的一个瓶颈。技术实现要素:本发明旨在至少在一定程度上解决相关技术中的技术问题之一。为此,本发明的一个目的在于提出一种对目标区域基因突变进行富集和检测的方法,该方法通过引入额外碱基错配的阻滞物进行阻滞取代扩增,达到富集检测目标突变的目的。现在市面存在的一些ctdna检测方法,都不太能满足癌症术后监控和癌症早期筛查的需求。ngs检测虽然通量高,但检测灵敏度不太高;数字pcr检测虽然检测灵敏度很高,但通量较差,一次只能检测一个突变位点;基于arms原理的qpcr检测的突变类型比较有限,灵敏度和通量都不算太高。2017年报道的一种名为“阻滞取代扩增”的pcr技术(blockerdisplacementamplification,bda),能够达到<0.1%突变频率样本的检测要求,并能同时完成156种突变序列的检测。bda方法通过加入不同的阻滞扩增序列能够同时将多种不同的低频突变dna序列富集到一个较高的频率,最后通过一代测序技术对富集后的dna序列进行测序。bda方法不仅能够检测ctdna中的低频突变dna,还因其依托于一代测序技术使其能达到极高的检测准确性,这种兼顾了检测的高灵敏度、高准确度以及一定通量的检测方法非常适用于癌症术后监控和癌症早期筛查。而且,与ngs检测和基于arms原理的qpcr检测方法相比,bda检测方法拥有较高灵敏度和准确度,其依托于一代测序,而一代测序一直是测序的金标准,可以达到99.9%的准确度。可以将其用于癌症的术后监控和癌症的早期筛查。然而发明人在研究过程中发现bda方法也存在着较为严重的缺陷,例如在对阳性样本和阴性样本进行富集检测时发现原本的序列在富集扩增过程中出现了一两个碱基错误,并且这种序列扩增的错误不能通过增加酶的保真性得以解决。出现这种序列扩增错误可能是阻滞取代扩增体系的不稳定性导致的。发生扩增错误的碱基位置存在于待扩增序列与特异性阻滞扩增序列碱基互补的区域内,但不是固定发生在某个碱基位置。有时发生在待检测碱基位点附近,有时正好发生在待检测位点上。这对后续正确检测造成了十分重要的影响,如果不能解决这个问题,将不能保证bda方法检测的准确性。本发明着重于解决在bda方法检测中出现的上述扩增序列错误的问题,在保证较高灵敏度的同时提高该方法的准确度,为推动该方法实现临床应用提供一些帮助。发明人研究发现,无论是改变pcr酶、pcr的退火延伸温度、引物与阻滞扩增序列的比例等因素均不能从根本上解决bda方法检测中出现的扩增序列错误的问题。发明人通过研究发现,以对阻滞扩增序列进行改造,可以完美的解决这个问题,既保证了该方法对于低频突变的检测能力,又解决了样本检测中出现的碱基突变的问题。这种优化的阻滞扩增序列是在原有的阻滞扩增序列基础上人为的引入一个或多个碱基的突变(与待检测目标区域位点碱基互补位点除外),这样的阻滞扩增序列无论是与野生型核酸样本,还是和突变型核酸样本均不能够完全碱基互补,但阻滞扩增序列与野生型核酸样本的结合力仍然大于与突变型核酸样本的结合力。具体而言,本发明提供了如下技术方案:在本发明的第一方面,本发明提供了一种对目标区域基因突变进行富集的方法,包括:(1)将含有待检测目标区域的核酸样本和第一引物、第二引物、阻滞扩增序列混合,进行第一pcr扩增,以便获得第一扩增产物;(2)基于所述第一扩增产物,利用所述第一引物和第三引物进行第二pcr扩增,以便获得第二扩增产物;其中,所述阻滞扩增序列的3’末端含有阻滞基团,所述阻滞扩增序列和所述第一引物含有部分重叠序列,所述待检测目标区域位于所述阻滞扩增序列与所述核酸样本的匹配区域内,所述第一引物的3’末端位于所述待检测目标区域的上游,所述阻滞扩增序列与所述核酸样本之间存在至少一个碱基不匹配位点,所述至少一个碱基不匹配位点不同于所述待检测目标区域的位点。本发明所提供的对目标区域基因突变进行富集的方法,其优化了阻滞取代扩增的pcr技术,在对含有待检测目标区域的核酸样本进行扩增时,加入第一引物和第二引物作为引物序列,同时加入阻滞扩增序列,阻滞扩增序列的3’末端含有阻滞基团,所设计的阻滞扩增序列与核酸样本之间存在至少一个碱基不匹配位点,该位点不同于所述待检测目标区域的位点。通过引入至少一个碱基不匹配位点,能够使得阻滞扩增序列既不与野生型核酸样本完全匹配,也不与突变型核酸样本进行完全匹配,但是阻滞扩增序列与野生型核酸样本的结合力仍然大于与突变型核酸样本的结合力。将阻滞扩增序列、第一引物和第二引物与核酸样本混合,扩增时,阻滞扩增序列竞争性与野生型核酸样本结合,使得野生型核酸样本的富集受到极大的抑制;同时第一引物与突变型核酸样本的结合能力大于阻滞扩增序列与突变型核酸样本的结合力,从而使得突变型核酸样本得到富集;由此实现对目标区域基因突变的富集,即可以保证对于低频突变的检测能力,又能够解决核酸样本检测过程中出现的碱基错误。根据本发明的实施例,以上所述的对目标区域基因突变进行富集的方法可以进一步包括如下技术特征:在本发明的一些实施例中,所述阻滞扩增序列与所述核酸样本之间存在2~5个碱基不匹配位点,优选存在2~3个碱基不匹配位点。由此可以解决核酸样本检测过程出现的碱基错误,提高对于低频突变检测的准确度。在本发明的一些实施例中,所述至少一个碱基不匹配位点的碱基类型为由c/g碱基突变为a/t碱基。在本发明的一些实施例中,所述阻滞扩增序列的tm值高于所述第一引物的tm值。在本发明的一些实施例中,所述阻滞扩增序列的tm值为60摄氏度~70摄氏度,优选为60摄氏度,所述第一引物的tm值为55摄氏度~60摄氏度。在本发明的一些实施例中,所述阻滞扩增序列去除所述部分重叠序列后序列的tm值高于所述第一引物去除所述部分重叠序列后序列的tm值。在本发明的一些实施例中,所述阻滞基团包括选自磷酸基团、间臂c3、间臂c5、倒置a、倒置g、倒置c、倒置t或者与所述待检测目标区域不匹配的核酸序列中的至少一种。在本发明的一些实施例中,所述阻滞扩增序列的碱基长度为10bp~40bp,所述第一引物的碱基长度为15bp~30bp,所述部分重叠序列的碱基长度为3~20bp。在本发明的一些实施例中,所述待检测目标区域基因突变频率在1%以下,所述第二扩增产物中待检测目标区域基因突变频率达到50%以上。在本发明的一些实施例中,所述第二扩增产物的丰度大于10%,指示所述待检测目标区域存在突变;所述第二扩增产物的丰度不超过10%,指示所述待检测目标区域不存在突变。在本发明的一些实施例中,所述核酸样本为循环肿瘤dna。在本发明的一些实施例中,所述基因突变包括选自碱基置换突变、移码突变、碱基缺失突变和碱基插入突变。在本发明的一些实施例中,所述目标区域为egfrg719s突变,所述第一引物为seqidno:1,所述第二引物为seqidno:2,所述阻滞扩增序列为seqidno:3,所述第三引物为seqidno:4。在本发明的第二方面,本发明提供了一种对目标区域基因突变进行检测的方法,包括:基于本发明第一方面任一实施例所述的方法对目标区域基因突变进行富集,以便获得富集产物;将所述富集产物进行建库、测序,基于所述测序结果,确定所述目标区域的基因突变;所述测序为高通量测序或者一代测序。附图说明图1是根据本发明的实施例提供的阻滞取代扩增的pcr技术的原理图。图2是根据本发明的实施例提供的在设计阻滞扩增序列时的示意图。图3是根据本发明的实施例提供的在利用第一引物、第二引物和阻滞扩增序列进行第一pcr扩增时的示意图。图4是根据本发明的实施例提供的在利用第一引物和第三引物进行第二pcr扩增时的示意图。图5是根据本发明的实施例1提供的测序结果图。图6是根据本发明的对比例1提供的测序结果图。具体实施方式下面详细描述本发明的实施例,需要说明的是所示出的实施例是示例性的,旨在用于解释本发明,而不能理解为对本发明的限制。阻滞取代扩增的pcr检测方法作为一种pcr扩增技术,具有较高的通量以及较高的灵敏度和准确度。其所具有较高的通量主要体现在以下几点:1、可以同时检测同一突变位点上的不同碱基突变类型以及相近的突变位点。例如可以通过加入同一种特异性阻滞扩增序列对kras的突变位点g12d、g12v、g12c、g12s、g12a、g13d进行同时富集,最后通过一代测序技术或者二代测序技术分别检测到突变。2、可以同时检测同种缺失突变的不同突变类型。例如可以通过加入同一种特异性阻滞扩增序列对egfr19del的多种突变类型进行同时富集,最后通过一代测序技术或者二代测序技术分别检测到突变。3、可以同时检测同种插入突变的不同突变类型。例如可以通过加入同一种特异性阻滞扩增序列对egfr20ins的多种突变类型进行同时富集,最后通过一代测序技术或者二代测序技术分别检测到突变。4、由于阻滞扩增序列的特殊设计使得不同的阻滞扩增序列可以在同一退火延伸温度60℃下进行富集,我们可以实现在一个反应体系中对多种不同的突变进行富集。例如可以将egfr的l858r、t790m、19del,kras的g12d、g12v、g12c、g12s、g12a、g13d,pik3cae545k、nrasq61k、brafv600e等通过在一个反应体系中加入对应的阻滞扩增序列同时富集。最后通过一代测序技术或者二代测序技术分别检测到突变。同时,其所具有的较高的灵敏度和准确度,与其原理是分不开的。如图1所示,从图中可以看出,当引物p(primerp)和阻滞扩增序列(blockerb)组合作用于野生型核酸样本时,primerp和blockerb竞争性结合到野生型核酸样本上,此时,blockerb的结合能力大于primerp,因此野生型核酸样本的扩增效率得到了极大得抑制;而当primerp和blockerb组合作用于突变型核酸样本时,由于突变型核酸样本上存在突变,blockerb与突变型核酸样本并非完全匹配,导致其结合能力弱于primerp,因此突变型核酸样本得到极大得扩增。依托于对于野生型核酸样本和突变型核酸样本不同的扩增效率,可以将原本丰度较低的突变型核酸样本进行富集。这样原本<1%突变频率的样本被富集到50%-90%的突变频率,检测就变得十分容易。由此该方法所具有的较高的灵敏度和准确率对癌症的术后监控和癌症的早期筛查来说是一种很好的检测方法。本发明通过对该方法中所用到的阻滞扩增序列进行优化设计,即在原有的阻滞扩增序列上人为的引入一个或多个碱基的突变,使得所引入的一个或多个碱基突变与核酸样本不能进行匹配(与待检测目标区域位点不同),这样的阻滞扩增序列与野生型核酸样本和突变型核酸样本均不完全碱基互补,但是阻滞扩增序列与野生型核酸样本的结合力仍然大于与突变型核酸样本的结合力。由此,可以完美的解决阻滞取代扩增体系不稳定的问题,完成对多种低频突变序列的同时富集检测,有望改善目前市场上缺乏对癌症的术后监控和癌症的早期筛查的ctdna检测技术的状况。在本发明的一些优选实施方式中,在阻滞扩增序列的设计上,还要遵循一些基本的设计原则,如图2所示,包括:1.阻滞扩增序列(blocker)与一条引物(primer)有部分区域重合,但不能完全重合,并且该引物序列不能包含待检测目标区域位点的碱基位置;2.待检测目标区域位点的碱基位置需在阻滞扩增序列与核酸样本(待扩增序列)结合的区域内,且不能在阻滞扩增序列与引物的重叠区域;3.阻滞扩增序列的tm值要高于引物的tm值;4.引物的tm值(tmp)最好在55℃—60℃范围内,阻滞扩增序列的tm值(tmb)最好在60℃—70℃范围内;5.阻滞扩增序列除去与引物重合区域的序列tm值(tm2)要高于引物除去与阻滞扩增序列重合区域的序列tm值(tm1);6.在阻滞扩增序列上人为引入突变碱基,在阻滞扩增序列与核酸样本之间引入碱基不匹配位点,所引入的突变碱基不能在待检测目标区域位点的碱基位置;7.在阻滞扩增序列上人为引入突变碱基,在阻滞扩增序列与核酸样本之间引入碱基不匹配位点,通常为c/g碱基转变为a/t碱基;8.在阻滞扩增序列上人为引入突变碱基,在阻滞扩增序列与核酸样本之间引入碱基不匹配位点,使得阻滞扩增序列的tm值降低,通常接近60℃。当然,在对阻滞取代扩增序列进行设计时,在阻滞扩增序列上人为引入突变碱基,在阻滞扩增序列与核酸样本之间引入碱基不匹配位点,所引入的突变碱基可以设计在阻滞扩增序列与引物序列的非重叠区域;也可以设计在阻滞扩增序列与引物序列的重叠区域。为此,在本发明的一个方面,本发明提供了一种对目标区域基因突变进行富集的方法,包括:(1)将含有待检测目标区域的核酸样本和第一引物、第二引物、阻滞扩增序列混合,进行第一pcr扩增,以便获得第一扩增产物;(2)基于所述第一扩增产物,利用所述第一引物和第三引物进行第二pcr扩增,以便获得第二扩增产物;其中,所述阻滞扩增序列的3’末端含有阻滞基团,所述阻滞扩增序列和所述第一引物含有部分重叠序列,所述待检测目标区域位于所述阻滞扩增序列与所述核酸样本的匹配区域内,所述第一引物的3’末端位于所述待检测目标区域的上游,所述阻滞扩增序列与所述核酸样本之间存在至少一个碱基不匹配位点,所述至少一个碱基不匹配位点不同于所述待检测目标区域位点。含有待检测目标区域的核酸样本中含有野生型核酸样本和突变型核酸样本,利用第一引物、第二引物和阻滞扩增序列对该含有待检测目标区域的核酸样本进行第一pcr扩增,如图3所示,由于阻滞扩增序列与第一引物的设计不同,所以对于野生型核酸样本和突变型核酸样本的扩增效率不同。然后再利用第一引物和第三引物对第一扩增产物进行第二pcr扩增,如图4所示,第三引物针对第一扩增产物设计,能够进一步提高扩增的准确性。下面将结合实施例对本发明的方案进行解释。本领域技术人员将会理解,下面的实施例仅用于说明本发明,而不应视为限定本发明的范围。实施例中未注明具体技术或条件的,按照本领域内的文献所描述的技术或条件或者按照产品说明书进行。所用试剂或仪器未注明生产厂商者,均为可以通过市购获得的常规产品。实施例1实施例1提供了一种对目标区域进行富集和检测方法,并以检测egfrg719s突变为例进行说明,所用到的样本dna为:horizon的野生型cfdna和1%突变型cfdna(指样本dna中含目标突变位点的突变序列占总序列的1%)。包括如下步骤:1、利用第一引物、第二引物和阻滞扩增序列进行第一pcr扩增。首先配制如下pcr体系:表1第一pcr体系其中,2xthermo酶mix来自于platinumsuperfipcrmastermix,厂家为thermofishierscientific,货号为00772786。按照如下pcr程序进行第一pcr扩增:所用到的引物序列和阻滞扩增序列均由擎科生物科技有限公司进行人工合成,其中所用到的第一引物、第二引物和阻滞扩增序列分别如下所示:第一引物:cttatacaccgtgccgaacg(seqidno:1);第二引物:cccagcttgtggagcctcttac(seqidno:2);阻滞扩增序列:cgaacgcaccgtagcccag(seqidno:3)。2、利用第一引物和第三引物进行第二pcr扩增。配制如下反应体系:表2第二pcr体系试剂体积上述第一扩增产物(μl)1μl第一引物(10μm)2μl第三引物(10μm)2μl酶mix(μl)45μl总体积(μl)50μl其中所用到的酶mix购自于擎科生物科技有限公司,货号为l0402。按照如下pcr程序进行:所用到的第三引物为:ccagtggagaagctcccaacc(seqidno:4)。将所获得的第二扩增产物进行一代测序,所获得的测序结果如图5所示。其中,egfrg719s野生型序列为:aaagtgctgggctccggtg(seqidno:5),egfrg719s突变型序列为:aaagtgctgagctccggtg(seqidno:6)。从测序结果可以看出,本发明所提供的方法对于egfrg719s突变检测不会发生检测错误。对比例1对比例1提供了一种对egfrg719s进行检测的方法,其与实施例1的不同之处在于,所用到的阻滞扩增序列为:cgaacgcaccggagcccag(seqidno:7),其它步骤与实施例1相同。其实验结果如图6所示。从图6可以看出,所获得的检测结果出现碱基错误(如图6中箭头所示)。由此,通过本发明所提供的方法对目标区域突变富集检测,可以避免核酸样本检测过程中出现的碱基错误,提高准确性。本文中,术语“第一”、“第二”仅用于描述目的,而不能理解为指示或暗示相对重要性或者隐含指明所指示的技术特征的数量。由此,限定有“第一”、“第二”的特征可以明示或者隐含地包括至少一个该特征。在本发明的描述中,“多个”的含义是至少两个,例如两个,三个等,除非另有明确具体的限定。在本说明书的描述中,参考术语“一个实施例”、“一些实施例”、“示例”、“具体示例”、或“一些示例”等的描述意指结合该实施例或示例描述的具体特征、结构、材料或者特点包含于本发明的至少一个实施例或示例中。在本说明书中,对上述术语的示意性表述不必针对的是相同的实施例或示例。而且,描述的具体特征、结构、材料或者特点可以在任一个或多个实施例或示例中以合适的方式结合。此外,在不相互矛盾的情况下,本领域的技术人员可以将本说明书中描述的不同实施例或示例以及不同实施例或示例的特征进行结合和组合。尽管上面已经示出和描述了本发明的实施例,可以理解的是,上述实施例是示例性的,不能理解为对本发明的限制,本领域的普通技术人员在本发明的范围内可以对上述实施例进行变化、修改、替换和变型。sequencelisting<110>杭州瑞普基因科技有限公司<120>基于引入额外碱基错配的阻滞物进行阻滞取代扩增富集检测目标突变的方法<130>pidc3194257<160>7<170>patentinversion3.5<210>1<211>20<212>dna<213>artificialsequence<220><223>第一引物<400>1cttatacaccgtgccgaacg20<210>2<211>22<212>dna<213>artificialsequence<220><223>第二引物<400>2cccagcttgtggagcctcttac22<210>3<211>19<212>dna<213>artificialsequence<220><223>阻滞扩增序列<400>3cgaacgcaccgtagcccag19<210>4<211>21<212>dna<213>artificialsequence<220><223>第三引物<400>4ccagtggagaagctcccaacc21<210>5<211>19<212>dna<213>artificialsequence<220><223>egfrg719s野生型序列<400>5aaagtgctgggctccggtg19<210>6<211>19<212>dna<213>artificialsequence<220><223>egfrg719s突变型序列<400>6aaagtgctgagctccggtg19<210>7<211>19<212>dna<213>artificialsequence<220><223>阻滞扩增序列<400>7cgaacgcaccggagcccag19当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1
- 核酸杂交的电化学检测法的制作方法
- 一种评价中草药对dna氧化损伤保护作用的方法
- 卷烟烟气导致dna氧化损伤产生8-羟基脱氧鸟苷的测定评价方法及其应用的制作方法
- 一种新的多肽——人dna错配修复蛋白15和编码这种多肽的多核苷酸的制作方法
- 一种新的多肽——人dna错配修复蛋白8.9和编码这种多肽的多核苷酸的制作方法
- 一种新的多肽——人dna错配修复蛋白9.4和编码这种多肽的多核苷酸的制作方法
- 一种新的多肽——人dna错配修复蛋白8和编码这种多肽的多核苷酸的制作方法
- 一种新的多肽——人dna错配修复基因蛋白13和编码这种多肽的多核苷酸的制作方法
- 一种新的多肽——人dna错配修复基因蛋白11和编码这种多肽的多核苷酸的制作方法
- 一种新的多肽——人dna错配修复蛋白12.98和编码这种多肽的多核苷酸的制作方法