一种降低核酸扩增复制滑移率的方法与流程
本发明涉及生物
技术领域:
,特别是涉及一种降低核酸扩增复制滑移率的方法。
背景技术:
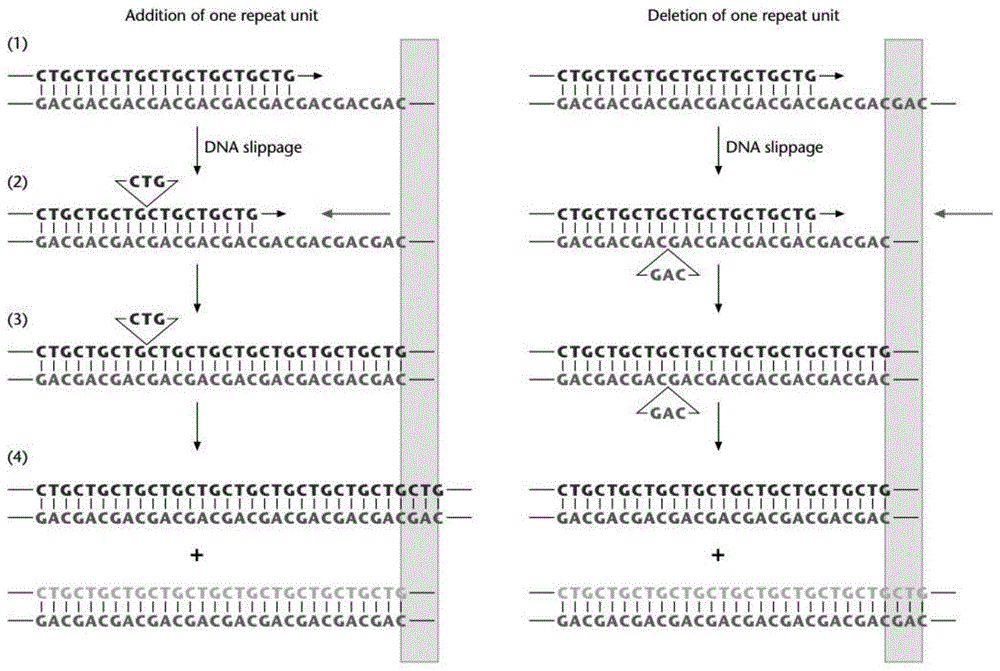
:所谓重复序列,就是在组成生物的dna中,重复出现的碱基序列。重复基因在生物基因组中广泛存在,如人类基因组中重复序列占比超过1/3。这些重复序列种类繁多,大多数还未确定其具体功能,但有研究已证明一些特定的重复序列在基因表达,调控和遗传等方面起着十分重要的作用。另外,重复序列是基因组不稳定的重要原因之一。基因组是动态结构,在生物体的生命过程中,基因会发生置换、移码、缺失、插入突变。重复序列由于其重复性高,在复制过程中极易产生插入或缺失重复单元,重复序列的再结合也可以导致染色体组的重组。现阶段对重复序列的检测主要采用基于pcr技术扩增片段长度多态性检测。此项技术中最严重的错误是扩增过程中发生复制滑移现象,即pcr过程中产生重复序列发生一个或几个重复单元的缺失或插入。dna复制过程中,因为子代链的插入或者模板链的脱落导致的引物-模板复合物在引物延伸的过程中发生错配,此时引物或者模板滑落导致一个重复单位形成碱基非配对环,这样导致其增加或减少一个或几个重复单位。这样的结构在扩增基因含短重复序列的pcr过程中较易出现。如果复制滑移片段比例较高,则无法区分目的片段因扩增效率不同而导致的目的片段扩增及复制滑移产物。这种结果直接导致无法对结果进行判别。因此,需要一种可降低复制滑移率的pcr方法,用以提高扩增片段长度多态性检测的分辨率,降低假阳性或无法判别的可能。技术实现要素:鉴于以上所述现有技术的缺点,本发明的目的在于提供一种降低核酸扩增复制滑移率的方法,用于解决现有技术中的问题。为实现上述目的及其他相关目的,本发明一方面提供一种降低核酸扩增复制滑移率的方法,包括:在sd聚合酶存在的条件下对模板核酸进行扩增,以提供扩增产物,所述模板核酸包括重复序列,所述重复序列包括多个重复单元,所述扩增产物的复制滑移率≤8%;复制滑移率=复制滑移片段峰面积/(目标片段峰面积+复制滑移片段峰面积)×100%。在本发明一些实施方式中,所述扩增产物的非特异性扩增率≤8%;非特异性扩增率=(复制滑移片段峰面积+其他非目的产物峰面积)/(复制滑移片段峰面积+其他非目的产物峰面积+目标片段峰面积)×100%。在本发明一些实施方式中,所述重复序列中,重复单元的长度为1~20个bp,优选为1~6个bp,更优选为3~5bp。在本发明一些实施方式中,所述重复单元选自atag。在本发明一些实施方式中,所述重复序列包括3~5个重复单元,模板核酸的长度不大于1000bp。在本发明一些实施方式中,所述对模板进行扩增中所使用的pcr体系包括:模板核酸、sd聚合酶、dntps。在本发明一些实施方式中,所述pcr体系还包括针对所述模板核酸的上游引物和下游引物。在本发明一些实施方式中,所述pcr体系还包括扩增促进剂,优选的,所述扩增促进剂选自甜菜碱、ssb、dmso中的一种或多种的组合。在本发明一些实施方式中,所述pcr体系还包括水性介质、缓冲液、镁离子中的一种或多种的组合。在本发明一些实施方式中,所述pcr体系中,模板核酸的浓度为2-200ng/μl,sd聚合酶的浓度为0.02-0.08u/μl,dntps的浓度为0.3-0.4mm,上游引物的浓度为0.3-0.6mm,下游引物的浓度为0.3-0.6mm,扩增促进剂的浓度为0.5-2.5m,镁离子的浓度为3-3.5mm。在本发明一些实施方式中,所述缓冲液选自sd聚合酶缓冲液。在本发明一些实施方式中,所述对模板进行扩增中,扩增进行30~40个循环,优选扩增30~35个循环,更优选扩增35个循环。本发明另一方面提供一种试剂盒,所述试剂盒用于所述的降低核酸扩增复制滑移率的方法。本发明另一方面提供一种pcr扩增体系,所述扩增体系包括:模板核酸、sd聚合酶、dntp,所述模板核酸包括重复序列,所述重复序列包括多个重复单元。附图说明图1显示为复制滑移产生的机制示意图。图2显示为本发明实施例2sd聚合酶扩增结果示意图。图3a显示为本发明对比例1phusion酶扩增结果示意图。图3b显示为本发明对比例22×taqplusmastermix(dyeplus)扩增结果示意图。图3c显示为本发明对比例3qhusion酶扩增结果示意图。图3d显示为本发明对比例4gloriaplus1standard扩增结果示意图。图3e显示为本发明对比例5gloriaplus1gc扩增结果示意图。具体实施方式为了使本发明的发明目的、技术方案和有益技术效果更加清晰,以下结合实施例对本发明进行进一步详细说明,熟悉此技术的人士可由本说明书所揭露的内容容易地了解本申请发明的其他优点及功效。本发明发明人经过大量研究,发现特定的酶的选择可以极大地提升针对重复序列的pcr反应的忠实性,从而可以降低重复序列在复制过程中产生的复制滑移率,并进一步发现配合特定的pcr体系,可以进一步改善非特异性扩增和复制滑移,从而提供了一种能够有效降低核酸扩增复制滑移率的核酸扩增方法,在此基础上完成了本发明。本发明第一方面提供一种降低核酸扩增复制滑移率的方法,包括:在sd聚合酶存在的条件下对模板核酸进行扩增,以提供扩增产物,所述模板核酸包括重复序列,所述重复序列包括多个重复单元。上述方法中,所述模板核酸通常可以包括目标扩增核酸序列,扩增中通常会使用一定的扩增体系对模板核酸进行扩增,所述扩增体系中通常包括sd聚合酶,即在sd聚合酶存在的条件下对模板核酸进行扩增,从而可以有效避免扩增产物出现非特异性扩增和复制滑移的现象。所述sd聚合酶通常是一种具有良好热稳定性的、人工合成的聚合酶,其可以通过各种市售渠道获得。本发明所提供的降低核酸扩增复制滑移率的方法中,扩增产物的复制滑移率可以被有效降低,通常来说,使用sd聚合酶对包括重复序列的模板核酸进行扩增时,扩增产物的复制滑移率通常可以≤8%,复制滑移率的计算方法具体如下:复制滑移率=复制滑移片段峰面积/(目标片段峰面积+复制滑移片段峰面积)×100%;其中,复制滑移片段峰面积指目标片段±10bp的扩增产物的峰面积,具体是指扩增产物中比目标片段长度短10bp和比目标片段长度长10bp的范围内、且除去目标片段的扩增产物在检测时所对应的峰面积的总和,这一部分扩增产物被认为是出现复制滑移的扩增产物;例如,当目标片段的长度为abp时,复制滑移片段峰面积则是长度为(a-10)~(a-1)bp、(a+1)~(a+10)bp的扩增产物的峰面积的总和,目标片段峰面积则是长度为abp的扩增产物的峰面积;当包括多个不同长度的目标片段时,例如,目标片段的长度为abp和bbp时,复制滑移片段峰面积则是长度为(a-10)~(a-1)bp、(a+1)~(a+10)bp、(b-10)~(b-1)bp、(b+1)~(b+10)bp的扩增产物的峰面积的总和,且上述范围内不包括扩增长度正确的扩增产物(例如,长度为abp和bbp的扩增产物)的峰面积,目标片段峰面积则是长度为abp和bbp的扩增产物的峰面积之和;目标片段峰面积则是扩增长度正确的扩增产物在检测时所对应的峰面积。合适的检测扩增产物并提供扩增产物峰面积的方法对于本领域技术人员来说应该是已知的,例如,毛细管电泳法、荧光检测毛细管电泳法、凝胶电泳法等。在本发明一具体实施例中,可以采用qsep100全自动核酸蛋白分析仪对扩增产物进行检测。本发明所提供的降低核酸扩增复制滑移率的方法中,扩增过程中同时可以具有良好的特异性,通常来说,使用sd聚合酶对包括重复序列的模板核酸进行扩增时,扩增产物的非特异性扩增率通常可以≤8%,非特异性扩增率的计算方法具体如下:非特异性扩增率=(复制滑移片段峰面积+其他非目的产物峰面积)/(复制滑移片段峰面积+其他非目的产物峰面积+目标片段峰面积)×100%;其中,复制滑移片段峰面积、目标片段峰面积的解释参照如上复制滑移率计算公式中的解释,其他非目的产物峰面积则为除复制滑移片段峰面积、目标片段峰面积以外,其他所有扩增产物的峰面积。本发明所提供的降低核酸扩增复制滑移率的方法中,所述方法通常用于针对包括重复序列的模板核酸,以提升扩增的忠实性。所述重复序列通常指在模板核酸或目标扩增核酸序列中重复出现的dna序列,本发明中所指的重复序列通常为串联重复序列,即指以相对恒定的短序列为重复单位,首尾相接串联连接形成的重复序列。所述包括重复序列的模板核酸中,重复单元的长度可以为1~20个bp、1~2个bp、2~3个bp、3~4个bp、4~5个bp、5~6个bp、6~8个bp、8~10个bp、10~12个bp、12~14个bp、14~16个bp、16~18个bp、或18~20个bp,优选为1~6个bp,更优选为3~5bp,在本发明一具体实施例中,重复单元的长度可以为3bp、4bp、或5bp。在本发明另一具体实施例中,所述重复单元可以选自atag等。所述包括重复序列的模板核酸中,重复序列可以包括3~5个重复单元,整体上的模板核酸的长度通常不大于1000bp。本发明所提供的降低核酸扩增复制滑移率的方法中,所使用的扩增体系通常需要包括各种核酸扩增所需要的试剂。例如,所述对模板进行扩增中所使用的pcr体系包括:模板核酸、sd聚合酶、dntp。所述dntp通常作为dna合成中的原料,具体可以包括datp、dgtp、dttp、dctp等。pcr体系中各组分的浓度对本领域技术人员来说是可以适当选择的,例如,模板核酸的起始量通常不小于20ng,模板核酸的浓度可以为2-200ng/μl、2-10ng/μl、10-30ng/μl、30-50ng/μl、50-100ng/μl、100-150ng/μl、或150-200ng/μl,浓度过低可进行浓缩;再例如,sd聚合酶的浓度可以为0.02-0.08u/μl、0.02-0.04u/μl、0.04-0.06u/μl、或0.06-0.08u/μl,即一个25μl体系加入0.5-2u酶,优选为0.035-0.045u/μl;再例如,dntp的浓度可以为0.3-0.4mm、0.3-0.35mm、或0.35-0.4mm,优选为0.37-0.38mm。所述pcr体系还可以包括针对所述模板核酸的上游引物和下游引物,所述上游引物和下游引物通常可以分别与目标扩增核酸序列的上游和下游在严格的条件下特异性杂交,上游引物和下游引物的具体序列通常取决于模板核酸,合适的引物序列的设计方法以及在pcr体系中的添加浓度对于本领域技术人员来说应该是已知的,例如,所述上游引物和/或下游引物的长度可以为15~50bp、15~25bp、25~35bp、或35~50bp;再例如,上游引物的浓度可以为0.3~0.6mm、0.3~0.4mm、0.4~0.5mm、或0.5~0.6mm,优选可以为0.4-0.5mm;再例如,下游引物的浓度可以为0.3~0.6mm、0.3~0.4mm、0.4~0.5mm、或0.5~0.6mm,优选可以为0.4-0.5mm。所述pcr体系还可以包括水性介质,所述水性介质通常可以用于调节pcr体系中各组分的浓度,通常可以作为稀释溶剂,合适的用于pcr体系的水性介质对于本领域技术人员来说应该是已知的,例如,所述水性介质可以是ddh2o等。所述pcr体系还可以包括缓冲液,所述缓冲液通常可以向pcr体系提供最合适酶催反应的条件,合适的用于pcr体系和/或对应sd聚合酶的缓冲液对于本领域技术人员来说应该是已知的,例如,所述缓冲液可以是sd聚合酶缓冲液等,具体可以是包括bsa的吐温-20缓冲液,其中,bsa的浓度可以为1-8μg/μl、1-2μg/μl、2-3μg/μl、3-4μg/μl、4-5μg/μl、5-6μg/μl、6-7μg/μl、或7-8μg/μl,ph可以为8.4~9.4、8.4~8.6、8.6~8.8、8.8~9.0、9.0~9.2、或9.2~9.4。所述pcr体系还可以包括镁离子,所述镁离子通常可以作为pcr体系中的催化剂,其可以与dntp形成dntp-mg与核酸骨架相互作用、并能影响polymerase的活性。合适的提供pcr体系中镁离子的方法和合适的pcr体系中镁离子的浓度对于本领域技术人员来说应该是已知的,例如,所述镁离子可以由溶解于pcr体系中的无机镁盐提供,再例如,所述无机镁盐可以是mgcl2等;再例如,镁离子的浓度可以为3~3.5mm,优选可以为3~3.2mm,更优选可以为3~3.1mm。本发明所提供的降低核酸扩增复制滑移率的方法中,所述扩增体系中还可以包括扩增促进剂,所述扩增体系中配合合适的扩增促进剂可以进一步改善非特异性扩增和复制滑移,例如,所述扩增促进剂可以选自甜菜碱、ssb、dmso等中的一种或多种的组合,再例如,所述扩增促进剂的浓度可以为0.5-2.5m、0.5-1m、1-1.5m、1.5-2m、或2-2.5m。本发明所提供的降低核酸扩增复制滑移率的方法中,扩增可以进行30~40个循环,优选可以扩增30~35个循环,更优选可以扩增35个循环。本发明所提供的降低核酸扩增复制滑移率的方法中,合适的pcr扩增条件对于本领域技术人员来说应该是已知的,例如,pcr扩增中,每一个循环通常包括dna变性、退火、延伸等步骤。再例如,还可以包括预变性、最后延伸等步骤。本发明第二方面提供sd聚合酶在降低核酸扩增复制滑移率中的用途。本发明第三方面提供一种试剂盒,所述试剂盒可以用于本发明第一方面所提供的降低核酸扩增复制滑移率的方法。所述试剂盒中,通常可以包括各种核酸扩增所需要的试剂,例如,可以包括sd聚合酶、dntp等,再例如,还可以包括如上所述的针对所述模板核酸的上游引物和下游引物、水性介质、缓冲液、提供镁离子的物质、扩增促进剂等中的一种或多种的组合。本发明第四方面提供一种pcr扩增体系,所述扩增体系包括:如上所述的模板核酸、sd聚合酶、dntp,所述模板核酸包括重复序列,所述重复序列包括多个重复单元。本发明所提供的pcr扩增体系中,还可以包括如上所述的针对所述模板核酸的上游引物和下游引物、水性介质、缓冲液、镁离子、扩增促进剂等中的一种或多种的组合。本发明所提供的降低核酸扩增复制滑移率的方法及其相关试剂盒、以及pcr扩增体系,能够有效减少核酸扩增中出现的非特异性扩增和复制滑移,其扩增获得的产物具有优良的扩增忠实性,具有良好的产业化前景。下面通过实施例对本申请的发明予以进一步说明,但并不因此而限制本申请的范围。除非另外说明,本发明中所公开的实验方法、检测方法、制备方法均采用本
技术领域:
常规的分子生物学、生物化学、染色质结构和分析、分析化学、细胞培养、重组dna技术及相关领域的常规技术。这些技术在现有文献中已有完善说明,具体可参见sambrook等molecularcloning:alaboratorymanual,secondedition,coldspringharborlaboratorypress,1989andthirdedition,2001;ausubel等,currentprotocolsinmolecularbiology,johnwiley&sons,newyork,1987andperiodicupdates;theseriesmethodsinenzymology,academicpress,sandiego;wolffe,chromatinstructureandfunction,thirdedition,academicpress,sandiego,1998;methodsinenzymology,vol.304,chromatin(p.m.wassarmananda.p.wolffe,eds.),academicpress,sandiego,1999;和methodsinmolecularbiology,vol.119,chromatinprotocols(p.b.becker,ed.)humanapress,totowa,1999等。实施例1对比例pcr体系中所使用的试剂具体信息如下:kapa2gfastmultiplexmix(kapabiosysterms),dsdnahsassaykit(thermofisherscientific),sdpolymerasereactionbufferincomplete(10×),mgcl2100mm,sdpolymeras10u/μl(生产商德国bioronlifescience),供应商为:广州爱丽丝生物技术有限公司。一:基因组定量使用dsdnahsassaykit或荧光定量pcr对基因组进行精确定量。具体定量流程参照dsdna定量方法说明书。本实施例选取的dna为外周血提取dna,经dsdna定量,浓度为:115.23ng/μl。按照不同要求可将dna进行稀释。二:靶区域扩增1、第一轮pcr扩增体系:采用0.2mlpcr管/8连管/96孔板,按照如下体系配置反应:表1ck组30μl体系panelmix4μldna(200ng)xμlkapa2gfastmultiplexmix15μlnuclease-freewaterto30μlpcr扩增程序具体如下:表22、pcr产物纯化1)pcr产物中加1.4倍体积ampurexpbeads(30μl体系加42μl)用移液器上下吹打,使回收产物与ampurexpbeads充分混匀。室温静置5-10分钟。用强力磁铁或磁力架吸附磁珠,直至溶液澄清为止。用移液器吸取上清,抛弃上清,保留磁珠。2)加入40ulbw13洗液,充分混匀。室温静置5分钟,用强力磁铁或磁力架吸附磁珠2分钟,直至溶液澄清为止。用移液器小心吸取上清,抛弃上清,保留磁珠。3)加入100μl80%乙醇,用磁力架反复在不同的两面来回吸附磁珠以充分悬浮磁珠便洗涤。4)用磁铁或磁力架吸附磁珠,直至溶液澄清为止。用移液器小心去除上清,避免吸到磁珠。5)室温放置,直至乙醇挥发干净。本步骤磁珠亦可放于50℃烘箱2分钟左右,快速蒸干乙醇。6)等待乙醇挥发期间,可准备下一步扩增反应试剂。3、第二轮pcr扩增体系:将在步骤2所得带有磁珠的tube管中加入以下pcr体系表3组分30μl体系pcrbarcodexxmix*2μlkapa2gfastmultiplexmix15μlnuclease-freewaterto30μl*不同的样本请使用不同的barcode引物pcr扩增程序具体如下:表44、pcr产物回收1)pcr产物中加1.0倍体积ampurexpbeads(30μl体系加30μl)用移液器上下吹打,使回收产物与ampurexpbeads充分混匀。室温静置5-10分钟。用强力磁铁或磁力架吸附磁珠,直至溶液澄清为止。用移液器吸取上清,抛弃上清,保留磁珠。2)加入40ulbw10洗液,充分混匀。室温静置5分钟,用强力磁铁或磁力架吸附磁珠2分钟,直至溶液澄清为止。用移液器小心吸取上清,抛弃上清,保留磁珠。3)加入100μl80%乙醇,用磁力架反复在不同的两面来回吸附磁珠以充分悬浮磁珠便洗涤。4)用磁铁或磁力架吸附磁珠,直至溶液澄清为止。用移液器小心去除上清,避免吸到磁珠。5)室温放置,直至乙醇挥发干净。本步骤磁珠亦可放于50℃烘箱2分钟左右,快速蒸干乙醇。6)加入20μlelutionbuffer,充分悬浮磁珠,室温静置2min以洗脱dna,将磁珠用磁铁吸附,所得到的上清dna溶液吸至新的1.5/0.5/0.2ml离心管中,elutionbuffer为10mmtris-hcl,ph8.0-8.5,也可以用te或超纯水替代。7)illumina平台nextseq500测序仪,pe150测序,上机数据量100m。使用sd酶进行pcr扩增,具体方法如下:一:基因组定量使用dsdnahsassaykit或荧光定量pcr对基因组进行精确定量。具体定量流程参照dsdna定量方法说明书。二:靶区域扩增1、第一轮pcr扩增体系:采用0.2mlpcr管/96孔pcr板,按照如下体系配置反应:表5pcr扩增程序具体如下:表62、pcr产物纯化1)pcr产物中加1.4倍体积ampurexpbeads(25μl体系加30μl)用移液器上下吹打,使回收产物与ampurexpbeads充分混匀。室温静置5-10分钟。用强力磁铁或磁力架吸附磁珠,直至溶液澄清为止。用移液器吸取上清,抛弃上清,保留磁珠。2)加入40μlbw13洗液,充分混匀。室温静置5分钟,用强力磁铁或磁力架吸附磁珠2分钟,直至溶液澄清为止。用移液器小心吸取上清,抛弃上清,保留磁珠。3)加入100μl80%乙醇,用磁力架反复在不同的两面来回吸附磁珠以充分悬浮磁珠便洗涤。4)用磁铁或磁力架吸附磁珠,直至溶液澄清为止。用移液器小心去除上清,避免吸到磁珠。5)室温放置,直至乙醇挥发干净。本步骤磁珠亦可放于50℃烘箱2分钟左右,快速蒸干乙醇。6)等待乙醇挥发期间,可准备下一步扩增反应试剂。3、第二轮pcr扩增体系:将在步骤2所得带有磁珠的tube管中加入以下pcr体系:表7组分30μl体系pcrbarcodexxmix*2μlkapa2gfastmultiplexmix15μlnuclease-freewaterto30μl*不同的样本请使用不同的barcode引物pcr扩增程序具体如下:表84、pcr产物回收1)pcr产物中加1.0倍体积ampurexpbeads(30μl体系加30μl)用移液器上下吹打,使回收产物与ampurexpbeads充分混匀。室温静置5-10分钟。用强力磁铁或磁力架吸附磁珠,直至溶液澄清为止。用移液器吸取上清,抛弃上清,保留磁珠。2)加入40ulbw10洗液,充分混匀。室温静置5分钟,用强力磁铁或磁力架吸附磁珠2分钟,直至溶液澄清为止。用移液器小心吸取上清,抛弃上清,保留磁珠。3)加入100μl80%乙醇,用磁力架反复在不同的两面来回吸附磁珠以充分悬浮磁珠便洗涤。4)用磁铁或磁力架吸附磁珠,直至溶液澄清为止。用移液器小心去除上清,避免吸到磁珠。5)室温放置,直至乙醇挥发干净。本步骤磁珠亦可放于50度烘箱2分钟左右,快速蒸干乙醇。6)加入20μlelutionbuffer,充分悬浮磁珠,室温静置2min以洗脱dna,将磁珠用磁铁吸附,所得到的上清dna溶液吸至新的1.5/0.5/0.2ml离心管中,elutionbuffer为10mmtris-hcl,ph8.0-8.5,也可以用te或超纯水替代。7)illumina平台nextseq500测序仪,pe150测序,上机数据量100m。测序引物panel选点具体如下:表9测序结果对比如表10所示:表10从测序结果看,使用sd酶与kapa2gfastmultiplexmix相比,sd酶的有效占比更高,为验证sd酶在str扩增中的优势。实施例2进一步以ms4为例,比较sd聚合酶与其他酶之间的扩增效果pcr体系中所使用的试剂具体信息如下:sdpolymerasereactionbufferincomplete(10×),mgcl2100mm,sdpolymeras10u/μl(生产商德国bioronlifescience),甜菜碱4m(生工生物),dntp10mm(生工生物)。上游引物:ms4f:actgcagtccaatctgggt(seqidno.21)下游引物:ms4r:atgaaatcaacagaggcttgc(seqidno.22)重复单元:atag,模板序列具体为:chr3:45582205-45582335131bpactgcagtccaatctgggtgacagagcaagaccctgtctcatagatagatagatagatagatagatagatagatagatagatagatagatagatagacag(seqidno.23),其中,重复序列具体为14个atag重复。pcr体系配置:pcr扩增模板起始量应不低于20ng,体系配置时推荐在冰盒上操作,配置完成后将该体系快速转移到92℃预热的pcr仪中,pcr扩增体系具体如表11所示:表11注意:温和混匀反应体系,如必要可以通过短暂快速离心将试剂收集到管底。pcr反应程序具体如表12所示:表12在pcr完成后可先进行琼脂糖凝胶电泳进行检测,观测到由明显条带则可进行下一步纯化。若无明显条带可增加5个循环。纯化:1.pcr产物中加1.0倍体积ampurexpbeads(25μl体系加25μl)用移液器上下吹打,使回收产物与ampurexpbeads充分混匀。室温静置5-10分钟。用强力磁铁或磁力架吸附磁珠,直至溶液澄清为止。用移液器吸取上清,抛弃上清,保留磁珠。2.加入40ulbw10洗液,充分混匀。室温静置5分钟,用强力磁铁或磁力架吸附磁珠2分钟,直至溶液澄清为止。用移液器小心吸取上清,抛弃上清,保留磁珠。3.加入100μl80%乙醇,用磁力架反复在不同的两面来回吸附磁珠以充分悬浮磁珠便洗涤。4.用磁铁或磁力架吸附磁珠,直至溶液澄清为止。用移液器小心去除上清,避免吸到磁珠。5.室温放置,直至乙醇挥发干净。本步骤磁珠亦可放于50度烘箱2分钟左右,快速蒸干乙醇。6.加入20μlelutionbuffer,充分悬浮磁珠,室温静置2min以洗脱dna。将磁珠用磁铁吸附,所得到的上清dna溶液吸至新的1.5/0.5/0.2ml离心管/96孔pcr管。elutionbuffer为10mmtris-hcl,ph8.0-8.5,也可以用te代替。qsep100检测:qsep100s1高分辨率卡夹使用前先室温平衡2h,根据操作说明装入机器,进行卡夹校正。将pcr产物进行qubit定量,取一定量的pcr产物加dilutionbuffer使其体积为10μl,浓度为1ng/μl。将配置好的待测溶液放入qsep100的相应孔位,选择方法1(吸样6kv,10s,片段长度鉴定8kv,200s),具体结果如图2所示。如图2所示,用sd酶添加1.5m甜菜碱的情况下,非特异性扩增和复制滑移均有良好的改善,复制滑移位置只占目的基因的7.94%。对比例1使用酶high-fidelitypcrmastermixwithhfbuffer生产商:newenglandbiolabs上游引物ms4f、下游引物ms4r、模板序列均同实施例2,pcr扩增体系具体如表13所示:表13pcr仪到达98℃时再将体系从冰上转到pcr仪上pcr反应参数具体如表14所示:表14pcr产物回收:1.pcr产物中加1.0倍体积ampurexpbeads(30μl体系加30μl)用移液器上下吹打,使回收产物与ampurexpbeads充分混匀。室温静置5-10分钟。用强力磁铁或磁力架吸附磁珠,直至溶液澄清为止。用移液器吸取上清,抛弃上清,保留磁珠。2.加入40ulbw10洗液,充分混匀。室温静置5分钟,用强力磁铁或磁力架吸附磁珠2分钟,直至溶液澄清为止。用移液器小心吸取上清,抛弃上清,保留磁珠。3.加入100μl80%乙醇,用磁力架反复在不同的两面来回吸附磁珠以充分悬浮磁珠便洗涤。4.用磁铁或磁力架吸附磁珠,直至溶液澄清为止。用移液器小心去除上清,避免吸到磁珠。5.室温放置,直至乙醇挥发干净。本步骤磁珠亦可放于50度烘箱2分钟左右,快速蒸干乙醇。6.加入20μlelutionbuffer,充分悬浮磁珠,室温静置2min以洗脱dna。将磁珠用磁铁吸附,所得到的上清dna溶液吸至新的1.5/0.5/0.2ml离心管/96孔pcr管。elutionbuffer为10mmtris-hcl,ph8.0-8.5,也可以用te代替。qsep100检测:qsep100s1高分辨率卡夹使用前先室温平衡2h,根据操作说明装入机器,进行卡夹校正。将pcr产物进行qubit定量,取一定量的pcr产物加dilutionbuffer使其体积为10μl,浓度至少为1ng/μl。将配置好的待测溶液放入qsep100的相应孔位,选择方法1(吸样6kv,10s,片段长度鉴定8kv,200s)。对比例2使用酶:2×taqmastermix(dyeplus),生产商:南京诺维赞生物科技有限公司上游引物ms4f、下游引物ms4r、模板序列均同实施例2,pcr扩增体系具体如表15所示:表15组分25μl体系ddh2o8.52×taqmastermix(dyeplus)12.5primer1(10μm)1primer2(10μm)1templatedna(20ng)2total25pcr反应参数具体如表16所示:表16pcr产物回收方法和qsep100检测方法参照对比例1。对比例3使用酶:qhusion生产商:启文生物上游引物ms4f、下游引物ms4r、模板序列均同实施例2,pcr扩增体系具体如表17所示:表17组分25μl体系2×qhusionpcrbuffer25μl10mmdntps2μl10μμprimerf1μl10μμprimerr1μltemplatedna(20ng)2μlqhusion(1.0u/μl)1μlnuclease-freewater25μlpcr仪到达98℃时再将体系从冰上转到pcr仪上pcr反应参数具体如表18所示:表18pcr产物回收方法和qsep100检测方法参照对比例1。对比例4使用酶:2×gloriaplus1standardpcrmix生产商:abclonaltechnology上游引物ms4f、下游引物ms4r、模板序列均同实施例2,pcr扩增体系具体如表19所示:表19组分25μl体系nuclease-freewater8.510μμprimerf1μl10μμprimerr1μltemplatedna(20ng)2μl2×gloriaplus1standardpcrmix12.5μltotal25μlpcr反应参数具体如表20所示:表20pcr产物回收方法和qsep100检测方法参照对比例1。对比例5使用酶:2×gloriaplus1gcpcrmix生产商:abclonaltechnology上游引物ms4f、下游引物ms4r、模板序列均同实施例2,pcr扩增体系具体如表21所示:表21组分25μl体系nuclease-freewater8.510μμprimerf1μl10μμprimerr1μltemplatedna(20ng)2μl2×gloriaplus1gcpcrmix12.5μltotal25μlpcr反应参数具体如表22所示:表22pcr产物回收方法和qsep100检测方法参照对比例1。各酶的扩增结果具体如图3所示。由图3可知phusionhigh-fidelitypcrmastermixwithhfbuffer(neb),qhusion酶(上海启文),2×taqplusmastermix(dyeplus)(vazyme)三种酶在对重复引物扩增时分别出现长度不同的非特异性扩增和无法确定pcr产物的涂抹带。各酶的复制滑移占比如表23所示,由表22可知,gloriaplus1standard,gloriaplus1gc,sd酶对减少重复序列扩增中的复制滑移均有有效减少,但gloriaplus1standard和gloriaplus1gc均产生微量非特异性扩增,效果相对于sd聚合酶仍有较大差距。表23复制滑移率=复制滑移片段峰面积/(目标片段峰面积+复制滑移片段峰面积)×100%注:复制滑移峰面积:目标片段±10bp峰面积(不包含目标片段)注:qhusion酶扩增产物为弥散状,未计入复制滑移。非特异性扩增率=(复制滑移片段峰面积+其他非目的产物峰面积)/(复制滑移片段峰面积+其他非目的产物峰面积+目标片段峰面积)×100%注:qhusion酶,phusionhigh-fidelitypcrmastermixwithhfbuffer,2×taqplusmastermix非特异性扩增过多,未计算非特异性扩增率。综上所述,本发明有效克服了现有技术中的种种缺点而具高度产业利用价值。上述实施例仅例示性说明本发明的原理及其功效,而非用于限制本发明。任何熟悉此技术的人士皆可在不违背本发明的精神及范畴下,对上述实施例进行修饰或改变。因此,举凡所属
技术领域:
中具有通常知识者在未脱离本发明所揭示的精神与技术思想下所完成的一切等效修饰或改变,仍应由本发明的权利要求所涵盖。序列表<110>上海韦翰斯生物医药科技有限公司<120>一种降低核酸扩增复制滑移率的方法<160>23<170>siposequencelisting1.0<210>1<211>27<212>dna<213>人工序列(artificialsequence)<400>1cagagactctgctaagcacatttccta27<210>2<211>29<212>dna<213>人工序列(artificialsequence)<400>2cagtattgtttagcacagaaacaggaagt29<210>3<211>24<212>dna<213>人工序列(artificialsequence)<400>3ggacacaggtccctgggagtagca24<210>4<211>27<212>dna<213>人工序列(artificialsequence)<400>4ggttggatgaggcaatagagatgaagt27<210>5<211>24<212>dna<213>人工序列(artificialsequence)<400>5cagtggaaagcaaaatggctgctc24<210>6<211>35<212>dna<213>人工序列(artificialsequence)<400>6ggtataatatagttagccagtaatccactggggct35<210>7<211>25<212>dna<213>人工序列(artificialsequence)<400>7cagaaagccaggtcccttcatagga25<210>8<211>27<212>dna<213>人工序列(artificialsequence)<400>8ccaggatacactgcaaatgctaagaag27<210>9<211>30<212>dna<213>人工序列(artificialsequence)<400>9ggaactggagtaaacaagattgctagatag30<210>10<211>26<212>dna<213>人工序列(artificialsequence)<400>10acatggcagcaggcatatacacatac26<210>11<211>28<212>dna<213>人工序列(artificialsequence)<400>11ggactttctagcctattggaaaggaaga28<210>12<211>32<212>dna<213>人工序列(artificialsequence)<400>12caatctctatttcttcagagcacttgtcagaa32<210>13<211>20<212>dna<213>人工序列(artificialsequence)<400>13gcctgggcgacagtaatggc20<210>14<211>31<212>dna<213>人工序列(artificialsequence)<400>14ccagagagacagaactgataggatctatgta31<210>15<211>27<212>dna<213>人工序列(artificialsequence)<400>15tcctacctttgttactctgcatgaaat27<210>16<211>29<212>dna<213>人工序列(artificialsequence)<400>16gcattccctatcctgtaatgaaagaaaga29<210>17<211>25<212>dna<213>人工序列(artificialsequence)<400>17caggtgttcactgcaagccatgcct25<210>18<211>32<212>dna<213>人工序列(artificialsequence)<400>18cccccaaaattctactgaagtaaaaagtttta32<210>19<211>27<212>dna<213>人工序列(artificialsequence)<400>19agcaagacaccatctcaagaaagaaaa27<210>20<211>38<212>dna<213>人工序列(artificialsequence)<400>20tgagaaattttacatttatgtttatgattctctttttt38<210>21<211>19<212>dna<213>人工序列(artificialsequence)<400>21actgcagtccaatctgggt19<210>22<211>21<212>dna<213>人工序列(artificialsequence)<400>22atgaaatcaacagaggcttgc21<210>23<211>100<212>dna<213>人工序列(artificialsequence)<400>23actgcagtccaatctgggtgacagagcaagaccctgtctcatagatagatagatagatag60atagatagatagatagatagatagatagatagatagacag100当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1
- 一种基于CRISPR‑Cas9系统的基因编辑载体及其应用的制造方法与工艺
- 一种基于CRISPR/Cas9技术制备水稻OsPIL15突变体的方法及应用与流程
- 检测黄曲霉毒素M1的核酸适配体、传感器、试剂盒及应用的制造方法与工艺
- 一组肌酸激酶同工酶核酸适配体及其应用的制造方法与工艺
- 用于抑制KRAS靶基因mRNA表达的寡核酸分子及其成套组合物的制造方法与工艺
- 用于抑制EIF4E靶基因mRNA表达的寡核酸分子及其成套组合物的制造方法与工艺
- 用于抑制VEGFA靶基因mRNA表达的寡核酸分子及其成套组合物的制造方法与工艺
- 一种新型向导RNA表达盒及在CRISPR/Cas系统中的应用的制造方法与工艺
- 一种具备双单链末端PCR产物的获得方法及其检测方法与流程
- 一种通过生物技术扩增脱氧胸苷三磷酸的方法与流程