水相中催化分子氧氧化生成2-氨基酚噁嗪-3-酮类化合物的方法与流程
本发明涉及有机杂环化合物合成技术领域,具体涉及一种水相中催化分子氧氧化生成2-氨基酚噁嗪-3-酮类化合物的方法。
背景技术:
酚噁嗪酮生物碱在自然界中分布广泛,被认为是一类独特的天然三环杂环化合物。研究发现,酚噁嗪酮类化合物具有广泛的药物特性,包括抗肿瘤、抗病毒、抗炎、抗菌、抗阿尔茨海默病等等。例如,放线菌素d(actinomycind)已被临床用于治疗多种类型的癌症。因此,药物化学研究常将酚噁嗪酮用作新药的母核结构。酚噁嗪酮也是天然染料和荧光探针的关键结构。而其中,2-氨基酚噁嗪-3-酮类化合物类化合物备受关注。
2-氨基酚噁嗪-3-酮类化合物的合成有很多方法,而由邻氨基苯酚类化合物氧化自偶联和交叉偶联反应生成2-氨基酚噁嗪-3-酮类化合物的方法最为简单高效(见式1)。许多氧化方法应用到此反应中,如酶促氧化[a)s.-j.yue,等,biotechnol.bioeng.2019,116,3072;b)j.bitzer等,j.antibiot.2006,59,86.],席夫碱金属络合物催化氧化[p.mahapatra等,inorg.chen.2017,56,5105.],电化学氧[t.l.williams等,acssus.chem.eng.2019,7,8979],双氧水氧化[j.kuehlborn等,acssust.chem.eng.2019,7,4414],多相锰催化氧化[s.ganguly等,newj.chem.2018,42,9517.],salenco催化氧化[k.maruyama等,chem.lett.1996,819.],铜络合物催化氧化[d.mondald等,inorg.chim.acta2019,486,719],无机氧化剂氧化[r.pasceri等,j.med.chem.2013,56,3310.],有机小分子催化氧化[g.szekely等,newj.chem.2015,39,5908.],仿酶催化氧化[y.zhou等,tetrahedronletters2014,55,2304]等。
这些方法使用的催化剂往往比较复杂,成本高,有些使用有机溶剂而导致后处理困难,难以避免含盐废水的产生,重要的是其交叉偶联反应的目标产物产率较低,从而限制了这些方法的使用。
技术实现要素:
本发明提出了一种水相中天然没食子酸和金属盐组合催化分子氧氧化邻氨基苯酚类化合物氧化自偶联和交叉偶联反应生成2-氨基酚噁嗪-3-酮类化合物的方法,该方法该法是在水相中反应,无需另外添加有机溶剂。
实现本发明的技术方案是:
一种水相中催化分子氧氧化生成2-氨基酚噁嗪-3-酮类化合物的方法,:以没食子酸为催化剂,金属盐为助催化剂,在氧气或空气环境中,使邻氨基苯酚类化合物在水相中、碱存在条件下反应生成2-氨基酚噁嗪-3-酮类化合物。
所述金属盐的金属为cu、fe、co、mn中的任意一种,酸根离子为醋酸根、碳酸根、盐酸根、硫酸根中的任意一种。金属盐为其中一种或两种以上任意组合。
所述碱为naoh、na2co3、nahco3、koh、k2co3或khco3中的任意一种。
所述邻氨基苯酚类化合物为邻氨基苯酚和取代的邻氨基苯酚。
所述催化剂没食子酸的用量为邻氨基苯酚类化合物物质的量的0.01-10%,所述助催化剂的用量为邻氨基苯酚类化合物物质的量的0.01-10%,所述水的用量为邻氨基苯酚类化合物质量的2-60倍;所述碱为邻氨基苯酚类化合物的0.05-1当量。
所述反应氧气分压为0.1-1.0mpa、温度为10-60℃、反应时间为2-50小时。
所述反应氧气分压为0.2-0.3mpa、温度为10-40℃、反应时间为4-20小时。
当邻氨基苯酚类化合物为两种组合时,交叉偶联反应时,两种不同的邻氨基苯酚类化合物的投料摩尔比为1:(0.9~1.2)。
所述合成2-氨基酚噁嗪-3-酮类化合物的方法,反应在水相中进行,无需添加其它有机溶剂。
本发明中,以没食子酸为催化剂,金属盐为助催化剂,直接投入使用。用于催化剂的没食子酸和助催化剂金属盐可以直接购买相应的化工产品。
本发明在使用过程中,反应效果随催化剂和助催化剂用量增加而提高,但催化剂用量增加生产成本也随之增加,过量的催化剂会带来分离困难。催化剂的用量为邻氨基苯酚类化合物质量的0.01-10%,优选0.03-2%。助催化剂的用量为邻氨基苯酚类化合物质的量的0.01-5%,优选0.03-1%。
本发明的方法在水相中进行,水的用量增加会降低反应液粘稠度而提高搅拌效果,进而提高反应效果,但水的用量过大会降低催化体系的浓度而降低反应效率,增加能源消耗。水的用量为邻氨基苯酚类化合物质量的2-60倍,优选10-40倍。
本发明中合成反应结束后,后处理工艺过程没有特别限定,产物分离提纯可以通过以下方法进行:氧化反应结束后,放置冷却,通过萃取、蒸馏,再经重结晶,得到产物。
本发明的有益效果是:反应在水相中进行,无需添加其它有机溶剂;催化剂简单,催化活性高,反应效率高;合成工艺简捷,废物少,环境友好,具有较强的工业应用前景。
附图说明
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
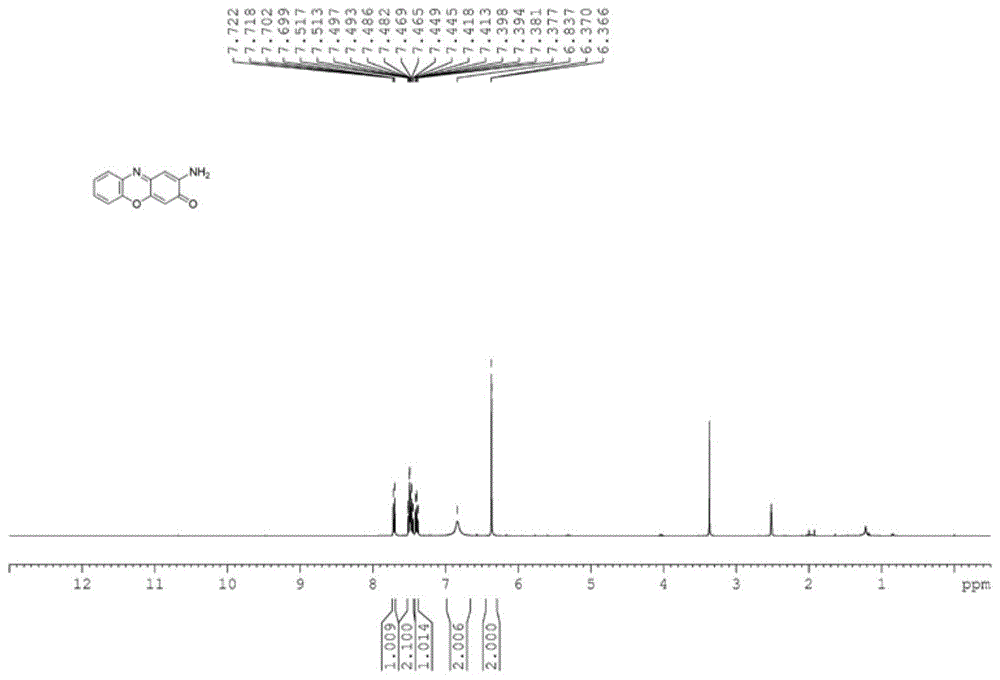
图1是实施例1制备的2-氨基酚噁嗪-3-酮的1hnmr谱图;
图2是实施例1制备的2-氨基酚噁嗪-3-酮的13hnmr谱图;
图3是实施例2制备的7-氯-2-氨基酚噁嗪-3-酮的1hnmr谱图;
图4是实施例2制备的7-氯-2-氨基酚噁嗪-3-酮的13hnmr谱图。
具体实施方式
下面将结合本发明实施例,对本发明的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有付出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
实施例1
2-氨基酚噁嗪-3-酮的合成:
在150ml的反应釜中,投入2.18g邻氨基苯酚、218mg没食子酸、2mg氯化钴、13mg硫酸锰、138mgk2co3和80ml水;搅拌下加热升温至15℃,通入氧气,保持反应釜内压强为1.0mpa,反应18小时后停止反应,冷却至室温,3×15ml乙酸乙酯萃取,合并乙酸乙酯层,旋蒸去除乙酸乙酯,剩余固体用异丙醇重结晶,抽滤,烘干,得黑色固体2.04g,产品经nmr(见附图1和2)、ms等方法确定结构为2-氨基酚噁嗪-3-酮,收率为94%,液相色谱仪分析产物纯度为97%。
实施例2
4-氯-2-氨基酚噁嗪-3-酮的合成:
在150ml的反应釜中,投入1.09g2-氨基苯酚、1.44g4-氯-2-氨基苯酚、14mg没食子酸、72mg氯化铜、0.67gkoh和65ml水;搅拌下保温25℃,压入空气,保持反应釜内压强为0.1mpa,反应4小时后停止反应,冷却至室温,3×15ml乙酸乙酯萃取,合并乙酸乙酯层,旋蒸去除乙酸乙酯,剩余固体用异丙醇重结晶,抽滤,烘干,得黑色固体2.2g,产品经nmr(见附图3和4)、ms等方法确定结构为7-氯-2-氨基酚噁嗪-3-酮,收率为87%,液相色谱仪分析产物纯度为97%。
按实施例1相同的方法合成其它2-氨基酚噁嗪-3-酮类化合物,其各种反应条件和反应结果见表1。
表1不同条件下合成各种2-氨基酚噁嗪-3-酮类化合物
以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!