一种含双(二苯胺)-四苯乙烯结构的二胺化合物及其制备方法、一种聚酰胺及其制备方法与流程
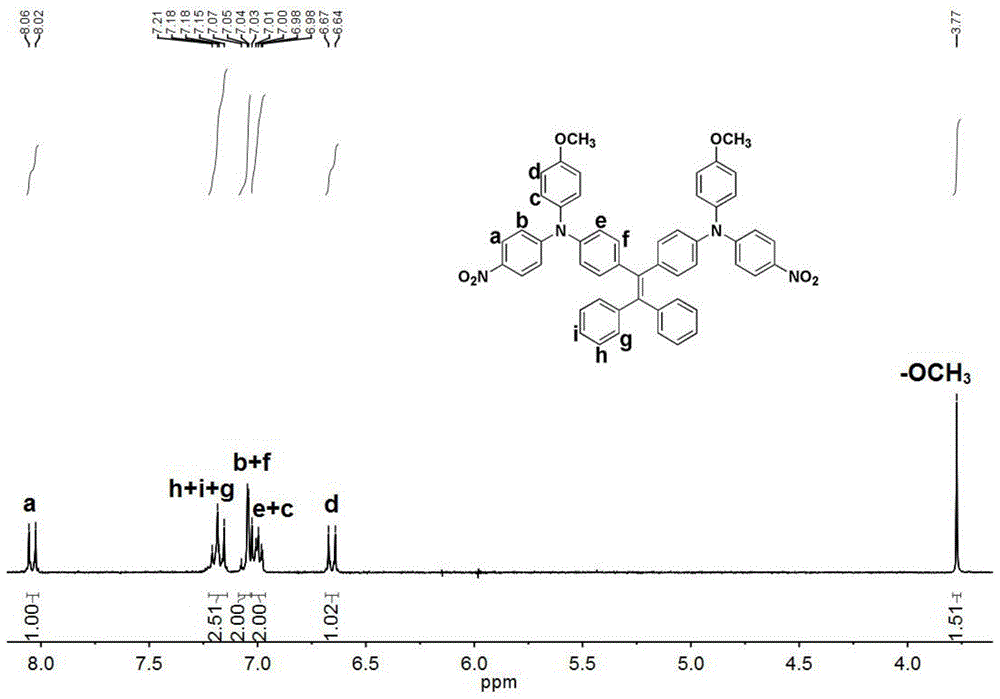
本发明涉及电控荧光
技术领域:
,尤其涉及一种含双(二苯胺)-四苯乙烯结构的二胺化合物及其制备方法、一种聚酰胺及其制备方法。
背景技术:
:电控荧光是指在外加电压的作用下材料发生可逆的荧光开关或变色现象,具有电控荧光性能的材料在传感器、监测电极表面、新型荧光显示器方面有很大的应用前景。相比于无机复合材料和小分子,聚合物由于加工性能好、结构易修饰而具有更大的优势。然而,目前合成的很多电控荧光聚合物在溶液态时能发出明亮荧光,在固态时则由于分子间强烈的π-π堆积效应使得荧光淬灭,即发生acq(aggregation-causedquenching)效应,这限制了它们在一些领域如有机发光二极管、荧光传感器等方面的应用。技术实现要素:本发明提供了一种含双(二苯胺)-四苯乙烯结构的二胺化合物,由本发明提供的二胺化合物制备得到的聚酰胺具有聚集态诱导发光性能,同时具有固态荧光量子产率高、氧化电位低、衰减速率慢、响应速度快、电控荧光循环稳定性能好、溶解性好、荧光开关时间短的特点。本发明提供了一种含双(二苯胺)-四苯乙烯结构的二胺化合物,具有式i所示结构:式i中:r为-ch3、-och3、-och2ch3、-n(ch3)2或-c(ch3)3;r1、r2、r3、r4、r5、r6独立地为-h、-ch3或-ch3(ch2)nch3,其中-ch3(ch2)nch3中n为1、2、3或4。优选的,所述含双(二苯胺)-四苯乙烯结构的二胺化合物具有式i-1~式i-5任一项所示结构:本发明还提供了上述技术方案所述含双(二苯胺)-四苯乙烯结构的二胺化合物的制备方法,包括以下步骤:(1)在保护气氛中,将4-卤代硝基苯、和碱性催化剂在极性有机溶剂中进行亲核取代反应,得到二苯胺衍生物;所述中r为-ch3、-och3、-och2ch3、-n(ch3)2或-c(ch3)3;所述二苯胺衍生物具有式ⅱ所示结构:(2)在保护气氛中,将所述步骤(1)得到的二苯胺衍生物、1,1-二苯基-2,2-二(4-卤代苯基)乙烯衍生物和催化剂在芳烃溶剂中进行偶联反应,得到二硝基化合物,所述二硝基化合物具有式iii所示结构:(3)将所述步骤(2)得到的二硝基化合物、pd/c催化剂和水合肼在有机溶剂中进行还原反应,得到式i所示含双(二苯胺)-四苯乙烯结构的二胺化合物。优选的,所述步骤(1)中碱性催化剂为三乙胺和/或三丙胺;所述亲核取代反应的温度为80~100℃,时间为30~45h。优选的,所述步骤(2)中1,1-二苯基-2,2-二(4-卤代苯基)乙烯衍生物包括1,1-二(2-甲基-3-乙基)苯基-2,2-二(4-氯苯基)乙烯或1,1-二(2-甲基-3-丙基)苯基-2,2-二(4-氯苯基)乙烯;所述催化剂包括主催化剂、助催化剂和相转移催化剂;所述主催化剂为铜和/或碘化亚铜,所述助催化剂为碳酸钾、碳酸钠、碳酸铯、氟化铯、氢氧化钠、氢氧化钾、氢化钾和氢化钠中的一种或多种,所述相转移催化剂为18-冠醚-6、15-冠醚-5、二苯并18-冠醚-6、苯并18-冠醚-6和苯并15-冠醚-5中的一种或多种。优选的,所述偶联反应的温度为140~180℃,时间为15~30h。优选的,所述还原反应的温度为85~90℃,时间为8~20h。本发明还提供了一种聚酰胺,所述聚酰胺具有式iv所示结构:其中-ar-具有式1~式5任一项所示结构:所述聚酰胺的聚合度为10~500。本发明还提供了上述技术方案所述聚酰胺的制备方法,包括以下步骤:在保护气氛中,将含双(二苯胺)-四苯乙烯结构的二胺化合物、二酸单体、缩合剂和助溶剂在n-甲基吡咯烷酮中混合,进行缩合反应,得到聚酰胺;所述含双(二苯胺)-四苯乙烯结构的二胺化合物为上述技术方案所述含双(二苯胺)-四苯乙烯结构的二胺化合物。优选的,所述缩合剂为亚磷酸三苯酯和吡啶;所述助溶剂为氯化钙;所述二酸单体包括对苯二甲酸、4,4'-联苯二甲酸、反式-1,4-环己烷二甲酸、4,4'-六氟丙烷对苯二甲酸和4,4'-磺酰基对苯二甲酸中的一种或多种。本发明提供了一种含双(二苯胺)-四苯乙烯结构的二胺化合物,所述二胺化合物具有式i所示结构,本发明提供的二胺化合物具有“2,2-三苯胺-四苯乙烯”结构,在由该二胺化合物制备的聚酰胺中,具有聚集诱导发射效应的四苯乙烯基团使聚酰胺在固态时具有明亮荧光的优异特性,并且使得聚酰胺具有较高的荧光量子产率;同时,聚酰胺中星型的四苯乙烯和三苯胺结构能够减弱聚酰胺分子链的密堆积、改善离子掺杂速率,进而缩短聚合物的电控荧光响应时间;此外,在三苯胺的对位引入推电子基团,可有效地降低氧化电位,改善聚酰胺的电化学稳定性。实验结果表明,本发明所提供的含2,2-三苯胺-四苯乙烯结构的二胺化合物可作为聚酰胺的单体,由其制备的聚酰胺不仅具有聚集诱导发射效应,还具有良好的溶解性、较短的响应时间、高荧光开关对比度和高度可逆的氧化还原过程。附图说明图1为实施例1制备得到的中间产物n,n'-双(4-硝基苯基)-n,n'-双(4-甲氧基苯基)-1,1-二苯基-2,2-二(4-苯胺)乙烯的氢核磁谱图;图2为实施例1制备得到的中间产物n,n'-双(4-硝基苯基)-n,n'-双(4-甲氧基苯基)-1,1-二苯基-2,2-二(4-苯胺)乙烯红外谱图;图3为实施例1制备得到的n,n'-双(4-氨基苯基)-n,n'-双(4-甲氧基苯基)-1,1-二苯基-2,2-二(4-苯胺)乙烯的氢核磁谱图;图4为实施例1制备得到的n,n'-双(4-氨基苯基)-n,n'-双(4-甲氧基苯基)-1,1-二苯基-2,2-二(4-苯胺)乙烯的h-hcosy谱图;图5为实施例1制备得到的n,n'-双(4-氨基苯基)-n,n'-双(4-甲氧基苯基)-1,1-二苯基-2,2-二(4-苯胺)乙烯的红外谱图;图6为应用例1制备得到的反式-1,4-环己烷二酸型聚酰胺的氢核磁谱图;图7为应用例1制备得到的反式-1,4-环己烷二酸型聚酰胺的红外谱图;图8为应用例1制备得到的反式-1,4-环己烷二酸型聚酰胺的循环伏安曲线图;图9为应用例1制备得到的反式-1,4-环己烷二酸型聚酰胺在不同电压下的电控荧光谱图;图10为应用例1制备得到的反式-1,4-环己烷二酸型聚酰胺在不同施加时间下的电控荧光响应谱图;图11为应用例1制备得到的反式-1,4-环己烷二酸型聚酰胺的电控荧光循环稳定性图。具体实施方式本发明提供了一种含双(二苯胺)-四苯乙烯结构的二胺化合物,具有式i所示结构:式i中:r为-ch3、-och3、-och2ch3、-n(ch3)2或-c(ch3)3;r1、r2、r3、r4、r5、r6独立地为-h、-ch3或-ch3(ch2)nch3,其中-ch3(ch2)nch3中n为1、2、3或4。在本发明中,所述含双(二苯胺)-四苯乙烯结构的二胺化合物优选具有式i-1~式i-5任一项所示结构:本发明还提供了上述技术方案所述含双(二苯胺)-四苯乙烯结构的二胺化合物的制备方法,包括以下步骤:(1)在保护气氛中,将4-卤代硝基苯、和碱性催化剂在极性有机溶剂中进行亲核取代反应,得到二苯胺衍生物;所述中r为-ch3、-och3、-och2ch3、-n(ch3)2或-c(ch3)3;所述二苯胺衍生物具有式ⅱ所示结构:(2)在保护气氛中,将所述步骤(1)得到的二苯胺衍生物、1,1-二苯基-2,2-二(4-卤代苯基)乙烯衍生物和催化剂在芳烃溶剂中进行偶联反应,得到二硝基化合物,所述二硝基化合物具有式iii所示结构:(3)将所述步骤(2)得到的二硝基化合物、pd/c催化剂和水合肼在有机溶剂中进行还原反应,得到式i所示含双(二苯胺)-四苯乙烯结构的二胺化合物。本发明在保护气氛中,将4-卤代硝基苯、和碱性催化剂在极性有机溶剂中进行亲核取代反应,得到二苯胺衍生物。在本发明中,所述4-卤代硝基苯优选包括对氟硝基苯或对氯硝基苯;在本发明中,所述中r为-ch3、-och3、-och2ch3、-n(ch3)2或-c(ch3)3;所述4-卤代硝基苯和的摩尔比优选为1:1.2~1.5。在本发明中,所述碱性催化剂优选包括三乙胺和/或三丙胺,所述4-卤代硝基苯、和碱性催化剂的摩尔比优选为1:1.2~1.5:1.2~1.5。在本发明中,所述极性有机溶剂优选包括二甲基亚砜。在本发明中,所述保护气氛优选为氮气。在本发明中,所述亲核取代反应的温度优选为80~100℃,进一步优选为85~95℃,时间优选为30~45h,进一步优选为35~40h。亲核取代反应完成后,本发明优选将亲核取代反应体系冷却至室温,然后将亲核取代反应体系倒入冰水中,充分混合后,过滤,收集固体,得到粗产物;将所述粗产物进行重结晶,得到式ⅱ所示二苯胺衍生物。在本发明中,所述重结晶的过程优选为:将粗产物溶于n,n-二甲基乙酰胺和乙醇的混合溶剂中,配制成热饱和溶液,所述n,n-二甲基乙酰胺和乙醇的体积比优选为1:2,所述热饱和溶液的温度优选为90℃;然后将所述热饱和溶液自然冷却至室温,过滤,收集固体,然后将所述固体进行真空干燥,所述真空干燥的温度优选为80℃,得到式ⅱ所示二苯胺衍生物。在本发明中,式ⅱ所示二苯胺衍生物中r基团与中的r基团相同。得到式ⅱ所示二苯胺衍生物后,本发明在保护气氛中,将所述二苯胺衍生物、1,1-二苯基-2,2-二(4-卤代苯基)乙烯衍生物和催化剂在芳烃溶剂中进行偶联反应,得到二硝基化合物,所述二硝基化合物具有式iii所示结构。在本发明中,所述保护气氛优选为氮气保护气氛。在本发明中,所述1,1-二苯基-2,2-二(4-卤代苯基)乙烯衍生物优选包括1,1-二(2-甲基-3-乙基)苯基-2,2-二(4-氯苯基)乙烯或1,1-二(2-甲基-3-丙基)苯基-2,2-二(4-氯苯基乙烯);所述催化剂优选包括主催化剂、助催化剂和相转移催化剂;所述主催化剂优选为铜和/或碘化亚铜,所述助催化剂优选为碳酸钾、碳酸钠、碳酸铯、氟化铯、氢氧化钠、氢氧化钾、氢化钾和氢化钠中的一种或多种,所述相转移催化剂优选为18-冠醚-6、15-冠醚-5、二苯并18-冠醚-6、苯并18-冠醚-6和苯并15-冠醚-5中的一种或多种。在本发明中,所述芳烃溶剂优选包括邻二氯苯、1,2,4-三氯苯、二氯甲苯和苯甲醚中的一种或多种。在本发明中,所述主催化剂、助催化剂和相转移催化剂的摩尔比优选为6~9:6~9:1。在本发明中,所述1,1-二苯基-2,2-二(4-卤代苯基)乙烯衍生物、二苯胺衍生物和催化剂的摩尔比优选为1:2~4:8~10,所述1,1-二苯基-2,2-二(4-卤代苯基)乙烯衍生物、二苯胺衍生物和催化剂的质量总和与芳烃溶剂的质量比优选为2~3:10。在本发明中,所述偶联反应的温度优选为140~180℃,进一步优选为160℃,时间优选为15~30h,进一步优选为24h。在本发明中,所述偶联反应的方程式如式(1)所示:偶联反应完成后,本发明优选将偶联反应体系趁热过滤,然后向滤液中加入乙醇,析出固体,过滤,收集固体,得到粗产物;将所述粗产物进行重结晶,得到式iii所示二硝基化合物。在本发明中,所述重结晶的过程优选为:将粗产物溶于n,n-二甲基乙酰胺和乙醇的混合溶剂中,配制成热饱和溶液,所述n,n-二甲基乙酰胺和乙醇的体积比优选为1:2,所述热饱和溶液的温度优选为90℃;然后将所述热饱和溶液自然冷却至室温,过滤,收集固体,然后将所述固体进行真空干燥,所述真空干燥的温度优选为80℃,得到式iii所示二硝基化合物。得到二硝基化合物后,本发明将所述二硝基化合物、pd/c催化剂和水合肼在有机溶剂中进行还原反应,得到式i所示含双(二苯胺)-四苯乙烯结构的二胺化合物。在本发明中,所述pd/c催化剂采用市售商品即可,所述pd/c催化剂中pd的质量分数优选为8~12%,更优选为10%。在本发明中,所述有机溶剂优选为二氧六环、乙醇和丙醇中的一种或多种。在本发明中,所述水合肼优选为水合肼水溶液,所述水合肼水溶液的质量浓度优选为80%。在本发明中,所述二硝基化合物与pd/c催化剂的质量比优选为1g:0.2~0.5g;二硝基化合物和水合肼的摩尔比优选为1:15~25,所述二硝基化合物的物质的量与有机溶剂的体积比优选为0.1~0.2mol:1l。在本发明中,所述二硝基化合物、pd/c催化剂、水合肼和有机溶剂的混合方式优选为:将二硝基化合物、pd/c催化剂和有机溶剂混合后,加热至还原反应温度,然后再与水合肼混合。在本发明中,所述水合肼优选采用滴加的方式,避免反应过于剧烈,出现危险。本发明对滴加的速率没有特别要求,只要使反应能够平稳进行即可。在本发明中,所述还原反应的温度优选为85~90℃,时间优选为8~20h,进一步优选为10~18h。在本发明中,所述还原反应的时间优选从开始滴加水合肼时,开始计时。还原反应完成后,本发明优选将还原反应体系趁热过滤,以除去pd/c催化剂,然后将所得滤液进行减压浓缩处理,得到浓缩液;再将所述浓缩液在氮气气氛下自然冷却至室温,析出固体,然后将所述固体进行真空干燥,得到式i所示含双(二苯胺)-四苯乙烯结构的二胺化合物。在本发明中,所述浓缩液的体积优选为减压浓缩前滤液体积的1/3~4/5,所述真空干燥的温度优选为80~100℃。本发明还提供了一种聚酰胺的制备方法,包括以下步骤:在保护气氛中,将含双(二苯胺)-四苯乙烯结构的二胺化合物、二酸单体、缩合剂和助溶剂在n-甲基吡咯烷酮中混合,进行缩合反应,得到聚酰胺;所述含双(二苯胺)-四苯乙烯结构的二胺化合物为上述技术方案所述含双(二苯胺)-四苯乙烯结构的二胺化合物。在本发明中,所述保护气氛优选为氮气气氛。在本发明中,所述二酸单体优选包括对苯二甲酸、4,4'-联苯二甲酸、反式-1,4-环己烷二甲酸、4,4'-六氟丙烷对苯二甲酸和4,4'-磺酰基对苯二甲酸中的一种或多种。在本发明中,所述缩合剂优选为亚磷酸三苯酯和吡啶。在本发明中,所述助溶剂优选为氯化钙。在本发明中,所述含双(二苯胺)-四苯乙烯结构的二胺化合物和二酸单体的摩尔比优选为1mmol:0.9~1.1mmol;所述含双(二苯胺)-四苯乙烯结构的二胺化合物、亚磷酸三苯酯和吡啶的用量比优选为1mmol:0.8~1.2ml:0.4~0.6ml;所述含双(二苯胺)-四苯乙烯结构的二胺化合物和助溶剂的用量比优选为1mmol:0.12g~0.18g;所述含双(二苯胺)-四苯乙烯结构的二胺化合物和n-甲基吡咯烷酮的用量比优选为1mmol:1.5~3ml。在本发明中,所述缩合反应的温度优选为160~180℃,时间优选为25~40h。本发明还提供了上述技术方案所述制备方法制备得到的聚酰胺,所述聚酰胺具有式iv所示结构:其中-ar-具有式1~式5任一项所示结构:所述聚酰胺的聚合度为10~500。在本发明中,所述式1~式5中结构式两端的“—”代表的是取代基位置,而不是代表“甲基”。本发明提供的聚酰胺含有“2,2-三苯胺-四苯乙烯”结构,使得聚酰胺具有固态荧光量子产率高、对比度高、氧化电位低、衰减速率慢、响应速度快、电控荧光循环稳定性能好、溶解性好、荧光开关时间短的特点。下面将结合本发明中的实施例,对本发明中的技术方案进行清楚、完整地描述。实施例1将40g(324.8mmol)的对甲氧基苯胺、35.2540g(249.9mmol)的对氟硝基苯、32.8672g(324.8mmol)的三乙胺和273ml的二甲基亚砜混合,在搅拌和氮气保护的条件下,于85℃反应36h;反应完成后,冷却至室温,出料于冰水中,充分搅拌后,过滤,得到粗产物;将粗产物用n,n-二甲基乙酰胺和乙醇(体积比为1:2)配制成热饱和溶液,热饱和溶液的温度为90℃,然后自然冷却至室温,过滤,将所得固体在80℃真空干燥,得到50.1g橙色的4-硝基-4'-甲氧基-二苯胺,产率为82.1%,4-硝基-4'-甲氧基-二苯胺的结构式如下所示:将40g(163.8mmol)的4-硝基-4'-甲氧基-二苯胺、34.65g(70.68mmol)1,1-二苯基-2,2-二(4-溴苯基)乙烯、36.19g(565.4mmol)铜粉、78.03g(565.4mmol)碳酸钾、18.68g(70.68mmol)18-冠醚-6和410ml的邻二氯苯混合,于160℃反应24h,反应结束后趁热过滤,向滤液中加入300ml乙醇,析出固体,然后过滤,得到粗产物;将所述粗产物用乙醇和n,n-二甲基甲酰胺(体积比为1:2)配制成热饱和溶液,热饱和溶液的温度为95℃,然后自然冷却至室温,过滤,将所得固体干燥,得到33.16g红色的n,n'-双(4-硝基苯基)-n,n'-双(4-甲氧基苯基)-1,1-二苯基-2,2-二(4-苯胺)乙烯,产率为57.43%,n,n'-双(4-硝基苯基)-n,n'-双(4-甲氧基苯基)-1,1-二苯基-2,2-二(4-苯胺)乙烯的结构式如下所示:将20g(24.48mmol)二(4-硝基苯基)-二(4'-甲氧基苯基)-4,4'-二氨基四苯乙烯、6gpd质量分数为10%的pd/c、100ml乙醇和100ml二氧六环混合,加热至85℃后,向溶液中缓慢滴加28.57ml质量分数为80%的水合肼溶液,滴加完成后,继续在85℃下搅拌10h,然后趁热过滤反应液除去pd/c,将所得滤液减压浓缩至原体积的1/3,氮气气氛下自然冷却至室温,然后将析出的固体在80℃真空干燥,得到黄色的n,n'-双(4-氨基苯基)-n,n'-双(4-甲氧基苯基)-1,1-二苯基-2,2-二(4-苯胺)乙烯粉末19.54g,产率为79.8%,n,n'-双(4-氨基苯基)-n,n'-双(4-甲氧基苯基)-1,1-二苯基-2,2-二(4-苯胺)乙烯的结构式如下所示:图1为实施例1制备得到的中间产物n,n'-双(4-硝基苯基)-n,n'-双(4-甲氧基苯基)-1,1-二苯基-2,2-二(4-苯胺)乙烯的氢核磁谱图;图2为实施例1制备得到的中间产物n,n’-双(4-硝基苯基)-n,n'-双(4-甲氧基苯基)-1,1-二苯基-2,2-二(4-苯胺)乙烯的红外谱图;图3为实施例1制备得到的n,n'-双(4-氨基苯基)-n,n'-双(4-甲氧基苯基)-1,1-二苯基-2,2-二(4-苯胺)乙烯的氢核磁谱图;图4为实施例1制备得到的n,n'-双(4-氨基苯基)-n,n'-双(4-甲氧基苯基)-1,1-二苯基-2,2-二(4-苯胺)乙烯的h-hcosy核磁谱图;图5为实施例1制备得到的n,n'-双(4-氨基苯基)-n,n'-双(4-甲氧基苯基)-1,1-二苯基-2,2-二(4-苯胺)乙烯的红外谱图;由图1核磁谱图可知,h原子的化学位移归属明确,可一一对应;由图2的红外谱图可知,图2中1314cm-1为-no2的对称振动吸收峰,1585cm-1为no2的非对称振动吸收峰,图5中3370cm-1,3460cm-1为-nh2的振动吸收峰。由图1~图5的红外和核磁结果可知,实施例1成功制备出了n,n'-双(4-硝基苯基)-n,n'-双(4-甲氧基苯基)-1,1-二苯基-2,2-二(4-苯胺)乙烯和n,n'-双(4-氨基苯基)-n,n'-双(4-甲氧基苯基)-1,1-二苯基-2,2-二(4-苯胺)乙烯。实施例2将45g(301.5mmol)的4-叔丁基苯胺、37.4966g(251.25mmol)的对氟硝基苯、30.5088g(301.5mmol)的三乙胺和308ml的二甲基亚砜混合,在搅拌和氮气保护的条件下,于85℃反应36h;反应完成后,冷却至室温,出料于冰水中,充分搅拌后,过滤,得到粗产物;将粗产物用n,n-二甲基乙酰胺和乙醇(体积比为1:2)配制成热饱和溶液,热饱和溶液的温度为90℃,然后自然冷却至室温,过滤,将所得固体在80℃真空干燥,得到58.0g橙色的4-硝基-4'-叔丁基-二苯胺,产率为85.4%,4-硝基-4'-叔丁基-二苯胺的结构式如下所示:将40g(147.0mmol)的4-硝基-4'-叔丁基-二苯胺、32.05g(65.37mmol)1,1-二苯基-2,2-二(4-溴苯基)乙烯、33.21g(523.0mmol)铜粉、20.92g(523.0mmol)氢氧化钠、23.56g(65.37mmol)二苯并18-冠醚-6和310ml的1,2,4-三氯苯混合;然后在氮气保护下,于160℃反应24h,反应结束后趁热过滤,向滤液中加入300ml乙醇,析出固体,然后过滤,得到粗产物;将所述粗产物用乙醇和n,n-二甲基甲酰胺(体积比为1:2)配制成热饱和溶液,热饱和溶液的温度为95℃,然后自然冷却至室温,过滤,将所得固体干燥,得到30.78g红色的n,n'-双(4-硝基苯基)-n,n'-双(4-叔丁基苯基)-1,1-二苯基-2,2-二(4-苯胺)乙烯,产率为54.18%,n,n'-双(4-硝基苯基)-n,n'-双(4-叔丁基苯基)-1,1-二苯基-2,2-二(4-苯胺)乙烯的结构式如下所示:将25g(28.77mmol)二(4-硝基苯基)-二(4'-叔丁基基苯基)-4,4'-二氨基四苯乙烯、7.5gpd质量分数为10%的pd/c、100ml乙醇和100ml二氧六环混合,加热至85℃后,向溶液中缓慢滴加31.18ml质量分数为80%的水合肼溶液,滴加完成后,继续在85℃下搅拌10h,然后趁热过滤反应液除去pd/c,将所得滤液减压浓缩至原体积的1/3,氮气气氛下自然冷却至室温,然后将析出的固体在80℃真空干燥,得到黄色的n,n'-双(4-氨基苯基)-n,n'-双(4-叔丁基苯基)-1,1-二苯基-2,2-二(4-苯胺)乙烯粉末19.11g,产率为82.1%,n,n'-双(4-氨基苯基)-n,n'-双(4-叔丁基苯基)-1,1-二苯基-2,2-二(4-苯胺)乙烯的结构式如下所示:实施例3将50g(367.2mmol)的n,n-二甲基-1,4-苯二胺、38.9577g(276.1mmol)的对氟硝基苯、37.1570g(367.2mmol)的三乙胺和383ml的二甲基亚砜混合,在搅拌和氮气保护的条件下,于85℃反应36h;反应完成后,冷却至室温,出料于冰水中,充分搅拌后,过滤,得到粗产物;将粗产物用n,n-二甲基乙酰胺和乙醇(体积比为1:2)配制成热饱和溶液,热饱和溶液的温度为90℃,然后自然冷却至室温,过滤,将所得固体在80℃真空干燥,得到57.2g橙色的4-硝基-4’-n,n’-二甲基二苯胺,产率为80.5%,其中4-硝基-4'-n,n'-二甲基二苯胺的结构式如下所示:将上述制备的40g(155.5mmol)的4-硝基-4'-n,n-二甲基二苯胺、33.08g(67.48mmol)1,1-二苯基-2,2-二(4-溴苯基)乙烯、38.56g(607.3mmol)铜粉、24.36g(607.3mmol)氢化钾、23.53g(87.7mmol)苯并15-冠醚-5和350ml的邻二氯苯混合;然后在氮气保护下,于160℃反应24h,反应结束后趁热过滤,向滤液中加入300ml乙醇,析出固体,然后过滤,得到粗产物;将所述粗产物用乙醇和n,n-二甲基甲酰胺(体积比为1:2)配制成热饱和溶液,热饱和溶液的温度为95℃,然后自然冷却至室温,过滤,将所得固体干燥,得到34.20g红色的n,n'-双(4-硝基苯基)-n,n'-双(4-n,n-二甲基苯基)-1,1-二苯基-2,2-二(4-苯胺)乙烯,产率为60.12%,n,n'-双(4-硝基苯基)-n,n'-双(4-n,n-二甲基苯基)-1,1-二苯基-2,2-二(4-苯胺)乙烯的结构式如下所示:将30g(35.59mmol)二(4-硝基苯基)-二(4'-n,n-二甲基苯基)-4,4'-二氨基四苯乙烯、9gpd质量分数为10%的pd/c、100ml乙醇和100ml二氧六环混合,加热至85℃后,向溶液中缓慢滴加33.83ml质量分数为80%的水合肼溶液,滴加完成后,继续在85℃下搅拌10h,然后趁热过滤反应液除去pd/c,将所得滤液减压浓缩至原体积的1/3,氮气气氛下自然冷却至室温,然后将析出的固体在80℃真空干燥,得到黄色的n,n'-双(4-氨基苯基)-n,n'-双(4-n,n-二甲基苯基)-1,1-二苯基-2,2-二(4-苯胺)乙烯粉末23.47g,产率为84.2%,n,n'-双(4-氨基苯基)-n,n'-双(4-n,n-二甲基苯基)-1,1-二苯基-2,2-二(4-苯胺)乙烯的结构式如下所示:实施例4将40g(291.6mmol)的4-乙氧基苯胺、32.7333g(210.43mmol)的对氯硝基苯、41.7775g(291.6mmol)的三丙胺和300ml的二甲基亚砜混合,在搅拌和氮气保护的条件下,于85℃反应36h;反应完成后,冷却至室温,出料于冰水中,充分搅拌后,过滤,得到粗产物;将粗产物用n,n-二甲基乙酰胺和丙醇(体积比为1:2)配制成热饱和溶液,热饱和溶液的温度为90℃,然后自然冷却至室温,过滤,将所得固体在80℃真空干燥,得到43.0g橙色的4-硝基-4'-乙氧基-二苯胺,产率为79.2%,4-硝基-4'-乙氧基-二苯胺的结构如下所示:将30g(116.2mmol)的4-硝基-4'-乙氧基-二苯胺、23.67g(48.76mmol)1,1-二(2-甲基-3-乙基)苯基-2,2-二(4-氯苯基)乙烯,81.71g(429.0mmol)碘化亚铜、46.8g(429.0mmol)碳酸钠、12.89g(58.53mmol)15-冠醚-5和380ml的二氯甲苯混合;然后在氮气保护下,于160℃反应24h,反应结束后趁热过滤,向滤液中加入350ml丙醇,析出固体,然后过滤,得到粗产物;将所述粗产物用丙醇和n,n-二甲基乙酰胺(体积比为1:2)配制成热饱和溶液,热饱和溶液的温度为90℃,然后自然冷却至室温,过滤,将所得固体干燥,得到22.74g红色的n,n'-双(4-硝基苯基)-n,n'-双(4-乙氧基苯基)-1,1-二(2-甲基-3-乙基)苯基-2,2-二(4-苯胺)乙烯,产率为50.19%,n,n'-双(4-硝基苯基)-n,n'-双(4-乙氧基苯基)-1,1-二(2-甲基-3-乙基)苯基-2,2-二(4-苯胺)乙烯的结构如下所示:将20g(21.53mmol)n,n'-双(4-硝基苯基)-n,n'-双(4-乙氧基苯基)-(2-(4-乙基-3-甲基苯基)-2-苯乙烯基二氯苯、6.0gpd质量分数为10%的pd/c、100ml丙醇和100ml二氧六环混合,加热至85℃后,向溶液中缓慢滴加17.34ml质量分数为80%的水合肼溶液,滴加完成后,继续在85℃下搅拌10h,然后趁热过滤反应液除去pd/c,将所得滤液减压浓缩至原体积的1/3,氮气气氛下自然冷却至室温,然后将析出的固体在80℃真空干燥,得到黄色的n,n'-双(4-氨基苯基)-n,n'-双(4-乙氧基苯基)-1,1-二(2-甲基-3-乙基)苯基-2,2-二(4-苯胺)乙烯粉末14.80g,产率为79.1%,n,n'-双(4-氨基苯基)-n,n'-双(4-乙氧基苯基)-1,1-二(2-甲基-3-乙基)苯基-2,2-二(4-苯胺)乙烯的结构如下所示:实施例5将35g(326.6mmol)的4-甲基苯胺、40.6448g(261.29mmol)的对氯硝基苯、46.7920g(326.6mmol)的三丙胺和330ml的二甲基亚砜混合,在搅拌和氮气保护的条件下,于85℃反应36h;反应完成后,冷却至室温,出料于冰水中,充分搅拌后,过滤,得到粗产物;将粗产物用n,n-二甲基甲酰胺和甲醇(体积比为1:2)配制成热饱和溶液,热饱和溶液的温度为95℃,然后自然冷却至室温,过滤,将所得固体在80℃真空干燥,得到50.9g橙色的4-硝基-4'-甲基-二苯胺,产率为85.4%,4-硝基-4'-甲基-二苯胺的结构如下式所示:将45g(116.2mmol)的4-硝基-4'-甲基-二苯胺、23.09g(44.96mmol)1,1-二(2-甲基-3-丙基)苯基-2,2-二(4-氯苯基)乙烯、74.29g(390.1mmol)碘化亚铜、59.3g(390.1mmol)氟化铯、13.76g(62.49mmol)15-冠醚-5和590ml的苯甲醚混合;然后在氮气保护下,于160℃反应24h,反应结束后趁热过滤,向滤液中加入300ml乙醇,析出固体,然后过滤,得到粗产物;将所述粗产物用甲醇和n,n-二甲基乙酰胺(体积比为1:2)配制成热饱和溶液,热饱和溶液的温度为95℃,然后自然冷却至室温,过滤,将所得固体干燥,得到23.26g红色的n,n’-双(4-硝基苯基)-n,n’-双(4-甲基苯基)-1,1-二(2-甲基-3-丙基)苯基-2,2-二(4-苯胺)乙烯,产率为57.67%,n,n'-双(4-硝基苯基)-n,n'-双(4-甲基苯基)-1,1-二(2-甲基-3-丙基)苯基-2,2-二(4-苯胺)乙烯的结构如下式所示:将20g(22.29mmol)n,n’-双(4-硝基苯基)-n,n'-双(4-乙氧基苯基)-1,1-二(2-甲基-3-丙基)苯基-2,2-二(4-苯胺)乙烯、6.0gpd质量分数为10%的pd/c、100ml丙醇和100ml二氧六环混合,加热至90℃后,向溶液中缓慢滴加17.34ml质量分数为80%的水合肼溶液,滴加完成后,继续在85℃下搅拌10h,然后趁热过滤反应液除去pd/c,将所得滤液减压浓缩至原体积的1/3,氮气气氛下自然冷却至室温,然后将析出的固体在80℃真空干燥,得到黄色的n,n'-双(4-氨基基苯基)-n,n'-双(4-甲基苯基)-1,1-二(2-甲基-3-丙基)苯基-2,2-二(4-苯胺)乙烯14.34g,产率为76.9%,n,n'-双(4-氨基基苯基)-n,n'-双(4-甲基苯基)-1,1-二(2-甲基-3-丙基)苯基-2,2-二(4-苯胺)乙烯的结构如下式所示:应用例1将0.7570g(1mmol)n,n'-双(4-氨基苯基)-n,n'-双(4-甲氧基苯基)-1,1-二苯基-2,2-二(4-苯胺)乙烯(由实施例1制备得到)、0.1722g(1mmol)反式-1,4-环己烷二酸,0.1287gcacl2、0.80ml亚磷酸三苯酯、0.40ml吡啶和1.5mln-甲基吡咯烷酮混合,在氮气气氛下,110℃反应3.5h;反应完成后,冷却至室温,出料至100ml乙醇中,然后过滤,得到黄色纤维状物;将所得黄色纤维状物质依次用乙醇、水、乙醇回流洗料30min,然后在真空烘箱于90℃烘干,得到甲氧基取代反式-1,4-环己烷二酸型聚酰胺,结构如下式所示,质量为0.6427g;应用例2将0.7570g(1mmol)n,n'-双(4-氨基苯基)-n,n'-双(4-甲氧基苯基)-1,1-二苯基-2,2-二(4-苯胺)乙烯(由实施例1制备得到)、0.1661g(1mmol)对苯二甲酸、0.1469gcacl2、1.1ml亚磷酸三苯酯、0.5ml吡啶和2.5mln-甲基吡咯烷酮混合;然后在氮气气氛下,110℃反应3.5h;反应完成后,冷却至室温,出料至100ml乙醇中,然后过滤,得到黄色纤维状产物;将所得黄色纤维状产物依次用乙醇、水、乙醇回流洗料30min,在真空烘箱于90℃烘干,得到甲氧基取代对苯二甲酸型聚酰胺,结构如下式所示,质量为0.8437g;应用例3将0.7570g(1mmol)n,n'-双(4-氨基苯基)-n,n'-双(4-甲氧基苯基)-1,1-二苯基-2,2-二(4-苯胺)乙烯(由实施例1制备得到)、0.2422g(1mmol)4,4’-联苯二甲酸、0.1785gcacl2、1.2ml亚磷酸三苯酯、0.6ml吡啶和2.6mln-甲基吡咯烷酮混合;然后在氮气气氛下,110℃反应3.5h;反应完成后,冷却至室温,出料至100ml乙醇中,然后过滤,得到黄色纤维状产物;将所得黄色纤维状产物依次用乙醇、水、乙醇回流洗料30min,在真空烘箱于90℃烘干,得到甲氧基取代4,4'-联苯二甲酸型聚酰胺,结构如下式所示,质量为0.9864g;应用例4将0.7570g(1mmol)n,n'-双(4-氨基苯基)-n,n'-双(4-甲氧基苯基)-1,1-二苯基-2,2-二(4-苯胺)乙烯(由实施例1制备得到)、0.3923g(1mmol)4,4’六氟丙烷对苯二甲酸、0.1693gcacl2、1.2ml亚磷酸三苯酯、0.6ml吡啶和3.0mln-甲基吡咯烷酮混合;然后在氮气气氛下,110℃反应3.5h;反应完成后,冷却至室温,出料至100ml乙醇中,然后过滤,得到黄色纤维状产物;将所得黄色纤维状产物依次用乙醇、水、乙醇回流洗料30min,在真空烘箱于90℃烘干,得到甲氧基取代4,4'-六氟丙烷对苯二甲酸型聚酰胺,结构如下式所示,质量为0.6497g;应用例5将0.7570g(1mmol)n,n'-双(4-氨基苯基)-n,n'-双(4-甲氧基苯基)-1,1-二苯基-2,2-二(4-苯胺)乙烯(由实施例1制备得到)、0.3063g(1mmol)4,4'-磺酰基对苯二甲酸、0.1473gcacl2、1.0ml亚磷酸三苯酯、0.5ml吡啶和3.0mln-甲基吡咯烷酮混合;然后在氮气气氛下,110℃反应3.5h;反应完成后,冷却至室温,出料至100ml乙醇中,然后过滤,得到黄色纤维状产物;将所得黄色纤维状产物依次用乙醇、水、乙醇回流洗料30min,在真空烘箱于90℃烘干,得到甲氧基取代4,4'-磺酰基对苯二甲酸型聚酰胺,结构式如下式所示,质量为0.6285g;应用例6将0.8091(1mmol)n,n'-双(4-氨基苯基)-n,n'-双(4-叔丁基苯基)-1,1-二苯基-2,2-二(4-苯胺)乙烯(由实施例2制备得到)、0.1722g(1mmol)反式-1,4-环己烷二酸、0.1697gcacl2、1.1ml亚磷酸三苯酯、0.5ml吡啶和2.5mln-甲基吡咯烷酮混合,在氮气气氛下,110℃反应3.5h;反应完成后,冷却至室温,出料至100ml乙醇中,然后过滤,得到黄色纤维状物;将所得黄色纤维状物质依次用乙醇、水、乙醇回流洗料30min,然后在真空烘箱于90℃烘干,得到叔丁基取代的反式-1,4-环己烷二酸型聚酰胺,结构式如下所示,质量为0.6079g;应用例7将0.8091(1mmol)n,n'-双(4-氨基苯基)-n,n'-双(4-叔丁基苯基)-1,1-二苯基-2,2-二(4-苯胺)乙烯(由实施例2制备得到)、0.1661g(1mmol)对苯二甲酸、0.1458gcacl2、1.2ml亚磷酸三苯酯、0.6ml吡啶和3.0mln-甲基吡咯烷酮混合,在氮气气氛下,110℃反应3.5h;反应完成后,冷却至室温,出料至100ml乙醇中,然后过滤,得到黄色纤维状物;将所得黄色纤维状物质依次用乙醇、水、乙醇回流洗料30min,然后在真空烘箱于90℃烘干,得到叔丁基取代的对苯二甲酸型聚酰胺,结构式如下所示,质量为0.7098g;应用例8将0.7830(1mmol)n,n'-双(4-氨基苯基)-n,n'-双(4-n,n-二甲基苯基)-1,1-二苯基-2,2-二(4-苯胺)乙烯(由实施例3制备得到)、0.3923g(1mmol)4,4'-六氟丙烷对苯二甲酸、0.1800gcacl2、1.0ml亚磷酸三苯酯、0.4ml吡啶和3.0mln-甲基吡咯烷酮混合,在氮气气氛下,110℃反应3.5h;反应完成后,冷却至室温,出料至100ml乙醇中,然后过滤,得到黄色纤维状物;将所得黄色纤维状物质依次用乙醇、水、乙醇回流洗料30min,然后在真空烘箱于90℃烘干,得到n,n二甲基基取代的4,4'-六氟丙烷对苯二甲酸型聚酰胺,结构如下式所示,质量为0.7539g;应用例9将0.7830(1mmol)n,n'-双(4-氨基苯基)-n,n'-双(4-n,n-二甲基苯基)-1,1-二苯基-2,2-二(4-苯胺)乙烯(由实施例3制备得到)、0.3063g(1mmol)4,4'-磺酰基对苯二甲酸、0.1587gcacl2、1.1ml亚磷酸三苯酯、0.4ml吡啶和1.9mln-甲基吡咯烷酮混合;然后在氮气气氛下,110℃反应3.5h;反应完成后,冷却至室温,出料至100ml乙醇中,然后过滤,得到黄色纤维状产物;将所得黄色纤维状产物依次用乙醇、水、乙醇回流洗料30min,在真空烘箱于90℃烘干,得到n,n-二甲基取代4,4'-磺酰基对苯二甲酸型聚酰胺,结构如下式所示,质量为0.6545g;应用例10将0.8271g(1mmol)n,n'-双(4-氨基苯基)-n,n'-双(4-乙氧基苯基)-1,1-二(2-甲基-3-乙基)苯基-2,2-二(4-苯胺)乙烯粉末(由实施例4制备得到)、0.2664g(1mmol)4,4'-联苯二甲酸、0.1685gcacl2、1.1ml亚磷酸三苯酯、0.6ml吡啶和2.8mln-甲基吡咯烷酮混合;然后在氮气气氛下,110℃反应3.5h;反应完成后,冷却至室温,出料至100ml乙醇中,然后过滤,得到黄色纤维状产物;将所得黄色纤维状产物依次用乙醇、水、乙醇回流洗料30min,在真空烘箱于90℃烘干,得到乙氧基取代4,4'-联苯二甲酸型聚酰胺,结构如下式所示,质量为0.7898g;应用例11将0.8081g(1mmol)n,n'-双(4-氨基基苯基)-n,n'-双(4-甲基苯基)-1,1-二(2-甲基-3-丙基)苯基-2,2-二(4-苯胺)乙烯(由实施例5制备得到)、0.1827g(1.1mmol)对苯二甲酸、0.1769gcacl2、1.0ml亚磷酸三苯酯、0.4ml吡啶和2.0mln-甲基吡咯烷酮混合;然后在氮气气氛下,110℃反应3.5h;反应完成后,冷却至室温,出料至100ml乙醇中,然后过滤,得到黄色纤维状产物;将所得黄色纤维状产物依次用乙醇、水、乙醇回流洗料30min,在真空烘箱于90℃烘干,得到甲基取代对苯二甲酸型聚酰胺,结构如下式所示,质量为0.7487g;结构表征应用例1所得产物甲氧基取代反式-1,4-环己烷二酸型聚酰胺的核磁谱图如图6所示,由图6可知,h原子的化学位移归属明确,可一一对应;应用例1所得甲氧基取代反式-1,4-环己烷二酸型聚酰胺的红外谱图如图7所示,由图7可知,3310cm-1为n-h伸缩振动吸收峰,1667cm-1为c=o伸缩振动吸收峰。由图6和图7可知,本发明应用例1所得甲氧基取代反式-1,4-环己烷二酸型聚酰胺的结构与预期结构相符。性能测试(1)荧光量子产率对应用例1制备得到的甲氧基取代反式-1,4-环己烷二酸型聚酰胺的荧光量子产率进行测试,测试方法为利用积分球测量聚酰胺的荧光量子产率。测试结果为:在n-甲基吡咯烷酮的稀溶液中荧光量子产率为0.85%,粉末状态(粉末的粒径为20~35微米)的荧光量子产率为19.60%,说明四苯乙烯结构的引入使聚合物具有聚集诱导发射特征,其中“双(二苯胺)-四苯乙烯”结构的引入使得聚酰胺在固态时具有高荧光强度。(2)循环伏安性能对应用例1制备得到的甲氧基取代反式-1,4-环己烷二酸型聚酰胺的循环伏安性能进行测试,测试方法如下:将聚酰胺溶解于n-甲基吡咯烷酮中,聚酰胺溶液的浓度为3mg/ml,将聚酰胺溶液滴涂于ito玻璃板上,90℃烘干5h,将得到的ito/聚合物膜作为工作电极,铂丝作为对电极,ag/agcl作为参比电极,0.1mol/l四丁基高氯酸铵(tbap)的乙腈溶液作为电解质;基于该三电极体系,通过电化学工作站进行循环伏安测试。测试应用例1所得甲氧基取代反式-1,4-环己烷二酸型聚酰胺的循环伏安曲线,结果如图8所示,由图8可知,该聚酰胺具有一对准可逆的氧化还原峰(氧化电位1.04v,还原电位0.49v),半波电位为0.77v,起始氧化电位为0.62v;200圈循环后曲线的电流值仅发生微小的衰减,说明应用例1制备得到的聚酰胺的电化学稳定性优异,证明了推电子甲氧基的引入可使材料氧化电位降低、循环稳定性增强。(3)电控荧光性能对应用例1所得甲氧基取代反式-1,4-环己烷二酸型聚酰胺的电控荧光性能进行测试,具体方法如下:将上述三电极体系搭建在荧光比色皿中,将荧光比色皿放置于荧光分光光度计中;通过电化学工作站调节电压,同时用荧光光谱仪监测其荧光强度的变化。应用例1所得甲氧基取代反式-1,4-环己烷二酸型聚酰胺的电控荧光谱图如图9所示,当外加电压从0v升至0.75v,523nm处的荧光强度逐渐降低,薄膜的荧光淬灭;反向施加电压时,薄膜的黄绿色荧光恢复,说明该聚酰胺具有可逆的电控荧光行为;最大发射峰处荧光开关对比度高达451,说明“双(二苯胺)-四苯乙烯”结构的引入使聚酰胺具有高荧光开关对比度。图10为所得甲氧基取代反式-1,4-环己烷二酸型聚酰胺在不同施加时间下的电控荧光曲线,方波电压为0~0.75v;逐渐缩短施加时间,其最高荧光强度逐渐减小,达到90%光学变化所需的时间逐渐减小,这是由于聚酰胺在短时间内发生的不完全的氧化还原反应;聚酰胺的荧光强度衰减速率较慢,当持续时间为20s时,荧光开/关时间较短(2s/0.32s),说明本发明提供的聚酰胺具有衰减速率慢和响应速度快的特点。这是因为星型的三苯胺和四苯乙烯结构能有效减弱分子链的密堆积,有利于电解质离子的嵌入与抽出,进而使聚酰胺具有衰减速率慢和响应速度快的特点。(4)电控荧光稳定性对应用例1制备得到的甲氧基取代反式-1,4-环己烷二酸型聚酰胺的电控荧光稳定性进行测试,测试方法为:在上述电控荧光测试体系中施加0.0-0.75v的方波电压,荧光光谱仪监测523nm处荧光的动态变化,结果如图11所示。由图11可知,当施加时间为20s时,经过100次转换后,荧光强度衰减程度约为12%,说明推电子甲氧基取代基的引入使聚合物具有优异的电控荧光循环稳定性。(5)溶解性能对应用例1制备得到的甲氧基取代反式-1,4-环己烷二酸型聚酰胺在常见有机溶剂中的溶解性能进行测试,测试方法为:在室温下,将10mg聚酰胺粉末溶于1ml溶剂中,室温下静置24h观察聚酰胺溶解程度,结果如表1所示:表1应用例1制备得到的甲氧基取代反式-1,4-环己烷二酸型聚酰胺的溶解性由表1测试结果可知,应用例1制备得到的甲氧基取代反式-1,4-环己烷二酸型聚酰胺在常见溶剂中的溶解性较好,有利于溶液加工法制备聚酰胺聚合物膜。(6)荧光开关时间对应用例1制备得到的甲氧基取代反式-1,4-环己烷二酸型聚酰胺的荧光开关时间进行测试,测试方法为:利用电化学工作站施加0-0.75v之间的方波电压,荧光光谱仪监测最大发射峰523nm处的荧光强度的变化,循环时间分别为5、10、20、30、40、60、80、100s时达到荧光对比度变化90%所需的荧光开/关时间如表2所示:表2应用例1制备得到的甲氧基取代反式-1,4-环己烷二酸型聚酰胺的荧光开关时间循环时间100s80s60s40s30s20s10s5s荧光关时间(s)0.240.280.280.280.320.320.320.28荧光开时间(s)14.289.488.005.043.002.000.760.34由表2可知,本发明制备得到的甲氧基取代反式-1,4-环己烷二酸型聚酰胺的荧光开关时间很短,说明了星型的三苯胺-四苯乙烯结构可以减弱分子链的堆积,加快电解质离子的传输速率,从而缩短荧光开关时间。本发明应用例2~11制备得到的聚酰胺也具有类似的性能,具有固态荧光量子产率高、对比度高、氧化电位低、衰减速率慢、响应速度快、电控荧光循环稳定性能好、溶解性好、荧光开关时间短的特点。综上,本发明提供的聚酰胺具有固态荧光量子产率高、对比度高、氧化电位低、响应速度快、电控荧光循环稳定性能好、溶解性好、荧光开关时间短的特点。以上所述仅是本发明的优选实施方式,应当指出,对于本
技术领域:
的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1