吲哚类生物碱、制备方法及作为降血糖药物的应用与流程
本发明涉及一种生物碱类化合物、制备方法及应用,尤其是一种吲哚类生物碱、制备方法及作为降血糖药物的应用。
背景技术:
中药吴茱萸为芸香科(rutaceae)吴茱萸属(tetradium)植物吴茱萸euodiarutaecarpa(juss.)benth.、石虎euodiarutaecarpa(juss.)benth.var.officinalis(dode)huang或疏毛吴茱萸euodiarutaecarpa(juss.)benth.var.bodinieri(dode)huang的干燥近成熟果实。根据《神农本草经》记载,吴茱萸具有味苦、辛温、有小毒等特性,归肝、脾、胃、肾经,具有助阳止泻、温中止痛、降逆止呕、散寒止痛等功,临床上常用于寒凝疼痛、胃寒呕吐、虚寒泄泻等症状的治疗。迄今为止没有以吴茱萸为原料,提取降血糖活性成份的相关报道。
技术实现要素:
本发明是为了解决现有技术所存在的上述技术问题,提供一种吲哚类生物碱、制备方法及作为降血糖药物的应用。
本发明的技术解决方案是:一种吲哚类生物碱,其特征在于结构式如下:
所述吲哚类生物碱的制备方法,其特征在于按照如下步骤进行:
a.将吴茱萸近成熟果实粉碎,用质量浓度70%的乙醇加热回流提取三次,合并提取液,减压浓缩并回收乙醇,得吴茱萸浸膏;将吴茱萸浸膏用水分散,依次用石油醚、乙酸乙酯、正丁醇多次等体积萃取,分别减压浓缩得石油醚萃取物、乙酸乙酯萃取物及正丁醇萃取物;
b.将乙酸乙酯萃取物进行硅胶柱层析,采用石油醚-乙酸乙酯按50:1,30:1,20:1,10:1,5:1,2:1,1:1,1:2梯度洗脱;
c.合并石油醚-乙酸乙酯20:1至5:1洗脱流份;
d.将所得洗脱流分经硅胶柱层析,用石油醚-乙酸乙酯按50:1,30:1,20:1,10:1,5:1,2:1,1:1,1:2梯度洗脱,每个梯度收集3个洗脱流份,每个洗脱流份为1l,将依次得到的24个洗脱流分分别记为n1~n24;
e.将洗脱流份n6经反相ods中压柱色谱分离,以甲醇-水梯度洗脱,流速15ml/min,洗脱6h,得到13个洗脱流份,每个洗脱流份为420ml,将依次得到的13个洗脱流份分别记为n6-1~n6-13;
f.将洗脱流份n6-10经反相半制备c18色谱柱hplc分离,甲醇:水=4:1,流速3.0ml/min,得到6个流份分别记为n6-10-1~n6-10-6;
g.将洗脱流份n6-10-3经反相半制备c18色谱柱hplc分离,甲醇:水=4:1,流速3.0ml/min,得单体化合物,即吲哚类生物碱。
所述的吲哚类生物碱作为降血糖药物的应用。
所述的吲哚类生物碱是作为抑制α-葡萄糖苷酶活性药物的应用。
本发明的吲哚类生物碱是从中药吴茱萸中提取,具有对α-葡萄糖苷酶活性抑制活性,可应用于制备降血糖药物。
附图说明
图1是本发明实施例化合物1的hresims谱图。
图2是本发明实施例化合物1的关键1h-1hcosy和hmbc相关图。
图3是本发明实施例化合物1的1hnmr谱图。
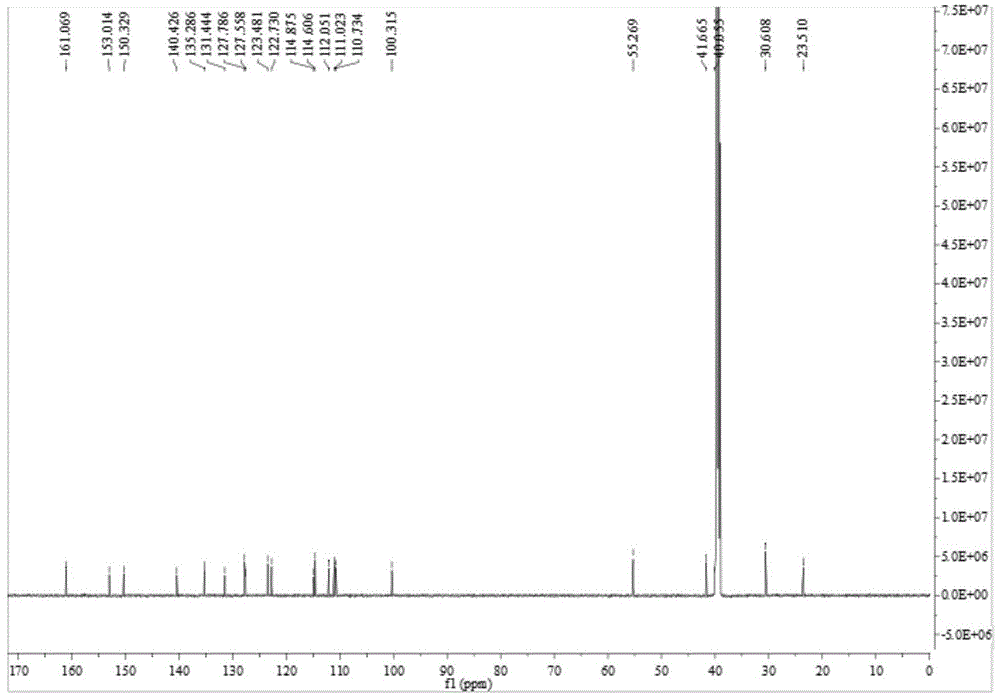
图4是本发明实施例化合物1的13cnmr谱图。
图5是本发明实施例化合物1的1h-1hcosy谱图。
图6是本发明实施例化合物1的hsqc谱图。
图7是本发明实施例化合物1的hmbc谱图。
具体实施方式
本发明的制备方法依次按照如下步骤进行:
a.将吴茱萸近成熟果实粉碎,用质量浓度70%的乙醇加热回流提取三次,合并提取液,减压浓缩并回收乙醇,得吴茱萸浸膏;将吴茱萸浸膏用水分散,依次用石油醚、乙酸乙酯、正丁醇多次等体积萃取,分别减压浓缩得石油醚萃取物、乙酸乙酯萃取物及正丁醇萃取物;
b.将乙酸乙酯萃取物进行硅胶柱层析,采用石油醚-乙酸乙酯按50:1,30:1,20:1,10:1,5:1,2:1,1:1,1:2梯度洗脱,每个梯度收集32l;
c.合并石油醚-乙酸乙酯20:1至5:1洗脱流份;
d.将所得洗脱流分经硅胶柱层析,用石油醚-乙酸乙酯按50:1,30:1,20:1,10:1,5:1,2:1,1:1,1:2梯度洗脱,每个梯度收集3个洗脱流份,每个洗脱流份为1l,将依次得到的24个洗脱流分分别记为n1~n24;
e.将洗脱流份n6经反相ods中压柱色谱分离,以甲醇-水梯度(10:90~90:10)洗脱,流速15ml/min,洗脱6h,得到13个洗脱流份,每个洗脱流份为420ml,将依次得到的13个洗脱流份分别记为n6-1~n6-13;
f.将洗脱流份n6-10经反相半制备c18色谱柱hplc分离,甲醇:水=4:1,流速3.0ml/min,得到6个流份分别记为n6-10-1~n6-10-6;
g.将洗脱流份n6-10-3经反相半制备c18色谱柱hplc分离,甲醇:水=4:1,流速3.0ml/min,得单体化合物1。
一.化合物1的结构测定:
化合物1为白色粉末。
化合物1的hresims谱图、h-1hcosy和hmbc相关谱图、1hnmr谱图、13cnmr谱图、1h-1hcosy谱图、hsqc谱图及hmbc谱图分别如图1~图7所示。
化合物1的核磁共振氢谱和碳谱数据如表1所示。
表1
注:13c-nmr(600mhz,dmso-d6),1h-nmr(150mhz,dmso-d6)
根据(+)-hr-esimsm/z372.1327[m+na]+(calcdforc20h19n3nao3,372.1319)结合核磁共振数据确定其分子式为c20h19n3o3(图1)。化合物1的1hnmr谱显示存在一个邻位二取代的苯环[δh7.47(d,j=8.4hz,h-16),7.79(td,j=8.4,1.2hz,h-17),7.32(td,j=7.8,1.2hz,h-18)和8.09(dd,j=7.8,1.2hz,h-19)],一个abx取代苯环[δh7.21(d,j=2.4hz,h-9),6.73(dd,j=8.4,2.4hz,h-11)和7.23(d,j=8.4hz,h-12)],一个烯氢信号[δh7.17(d,j=1.8hz,h-2)],两个脂肪族亚甲基信号[δh4.21(m,h-5)和2.95(m,h-6)],一个氮甲基信号[δh3.55(s,nch3-14)],一个甲氧基信号[δh3.76(s,och3-10)]以及一个可交换的质子[δh10.68(s,nh-1)]共振信号(表1)。13cnmr谱显示与以上基团相对应的碳信号外,还有四个sp2杂化的碳信号[δc123.5(c-2),150.3(c-3),110.7(c-7)和161.1(c-21)](表1)。根据以上数据推测该化合物具有一个吲哚环和一个喹唑酮环,利用2dnmr实验和图谱解析对其结构进行了进一步确定。通过hsqc谱解析,对nmr谱中氢原子及其相连的碳原子信号进行了准确归属。在1h-1hcosy谱中,同核质子偶合相关信号h-11/h-12,结合hmbc谱中,nh-1与c-7,c-8和c-12,h-9与c-7,c-11和c-13,h-2与c-8和c-13,h-12与c-8和c-10,och3-10与c-10的两键或三键异核远程相关信号以及它们的化学位移,表明化合物1的结构中存在c-10被甲氧基取代的吲哚单元(图2)。在1h-1hcosy谱中,同核质子偶合相关信号h-16/h-17/h-18/h-19,结合hmbc谱中,h-19与c-21和c-15,h-16与c-21和c-18,h-17与c-19,nch3-14与c-3和c-15的两键或三键异核远程相关信号以及它们的化学位移,表明化合物1的结构中存在n-14被甲基取代的喹唑酮单元(图2)。在1h-1hcosy谱中,同核质子偶合相关信号h-5/h-6,结合hmbc谱中,h-5与c-3,c-7和c-21,h-6与c-2和c-8的两键或三键异核远程相关信号以及它们的化学位移,表明化合物1结构中喹唑酮单元与吲哚单元分别连在c-5和c-6上(图2)。因此,综合以上波谱数据,确定了化合物1的结构式如下,为吲哚类生物碱。
二.本发明化合物1对α-葡萄糖苷酶活性抑制活性评价
采用96孔板筛选体系,先加80μl磷酸钾缓冲液(ph6.8),再加入α-glucosidase(0.1u/ml)10μl,加入不同浓度(10、20、30、50、100μm)药物(化合物1)溶液10μl,并设置空白对照,37℃恒温10min,然后加入pnpg(2.5mmol/l)20μl,37℃恒温反应15min,最后加入40μl的na2co3溶液(1mol/l)终止反应,于405mm波长下测定吸光度值(a值),并用graphpadprism6计算得到ic50值,ic50=23.92±0.81μm。
结果表明本发明的生物碱化合物1显示了对α-葡萄糖苷酶具有较强的抑制作用。
- 还没有人留言评论。精彩留言会获得点赞!