氨基吴茱萸碱衍生物及其制备方法与应用与流程
本发明属于吴茱萸碱技术领域,具体涉及氨基化吴茱萸碱及其制备方法与抗肿瘤应用。
背景技术:
左金丸出自《丹溪心法•火六》,由黄连、吴茱萸以6∶1比例组成,方中君药是性味苦寒的黄连,清肝火泻胃火,配伍性味辛热的吴茱萸,暖肝散寒开郁,疏肝理气,主治肝火犯胃证,左金丸在现代临床上主要用于治疗消化道溃疡、胃炎、胃食管反流等疾病。吴茱萸碱(evo)的合成方法有很多,报道最多的合成方法是以色胺和邻氨基苯甲酸为原料合成3,4-二氢-β-咔啉和n-甲基靛红酸酐,再将二者缩合得到吴茱萸碱的衍生物。通过使用不同取代基的原料,可以对evo进行结构修饰,得到不同位点、不同取代基的衍生物,这些改性对抗肿瘤活性有较大影响,取代基可以改变吴茱萸碱的表面性质以及电子性质,从而对改性分子的性能有重要影响,现有技术将含硼基团引入高活性吴茱萸碱衍生物的10位,得到了靶向衍生物13a,对hct116,mcf-7和a549三种肿瘤株的ic50值较低。文献研究表明结构修饰多集中在a环的10位,b环的13位,d环14位、e环的3位以及e环骨架的修饰,至今未见2位的修饰。
技术实现要素:
本发明公开了氨基化吴茱萸碱及其制备方法,用cck-8法进行细胞毒性实验,评价化合物对乳腺癌mda-mb-231细胞和结肠癌sw620细胞以及正常肝lo2细胞的抗增殖作用,用fitc/pi双染进行细胞凋亡的检测。
本发明采用如下技术方案:
氨基吴茱萸碱衍生物,具有如下化学结构式:
本发明公开了上述氨基吴茱萸碱衍生物的制备方法,包括以下步骤,将2-硝基吴茱萸碱衍生物还原,得到氨基吴茱萸碱衍生物;2-硝基吴茱萸碱衍生物具有如下化学结构式:
r1为氢、烷基或者烷氧基。
本发明中,还原为盐酸/氯化亚锡还原,现有技术用钯碳氢气还原,但是氢气属于易燃易爆气体,对生产安全不利。具体的,将氯化亚锡二水合物、溶剂、盐酸加入2-硝基吴茱萸碱衍生物中,室温~60℃反应15~25小时,然后用naoh调碱性至ph7~9,再用乙酸乙酯萃取,萃取液经过旋蒸蒸干溶剂,残留物用硅胶柱层析,得到氨基吴茱萸碱衍生物。
本发明公开了上述氨基化吴茱萸碱在制备药物尤其是抗肿瘤药物中的应用。
现有技术公开了一种吴茱萸碱衍生物的制备方法,以吴茱萸碱为原料,通过硝化、还原步骤,在10位成功引入氨基,制得硝基吴茱萸碱衍生物以及氨基吴茱萸碱衍生物,mtt法检测对h1688细胞的毒性,发现硝基吴茱萸碱衍生物的抗肿瘤效果更好。中药吴茱萸的干燥果实中的吴茱萸碱含量很少并且提取过程复杂,本发明通过化学方法从带有硝基的吴茱萸碱衍生物,在氯化亚锡/浓盐酸的还原条件下最终得到两种目标吴茱萸碱衍生物(2-nh2-evo、10-och3-2-nh2-evo),核磁共振氢谱、高分辨质谱红外光谱等的表征结果充分证实了化合物的成功合成。体外细胞毒性和凋亡实验表明,与吴茱萸碱相比,化合物2-nh2-evo对乳腺癌细胞mda-mb-231有很强的抑制作用,ic50值为0.79μm,同时能够显著诱导细胞凋亡,有望发展成为具有潜力的抗癌药物。
附图说明
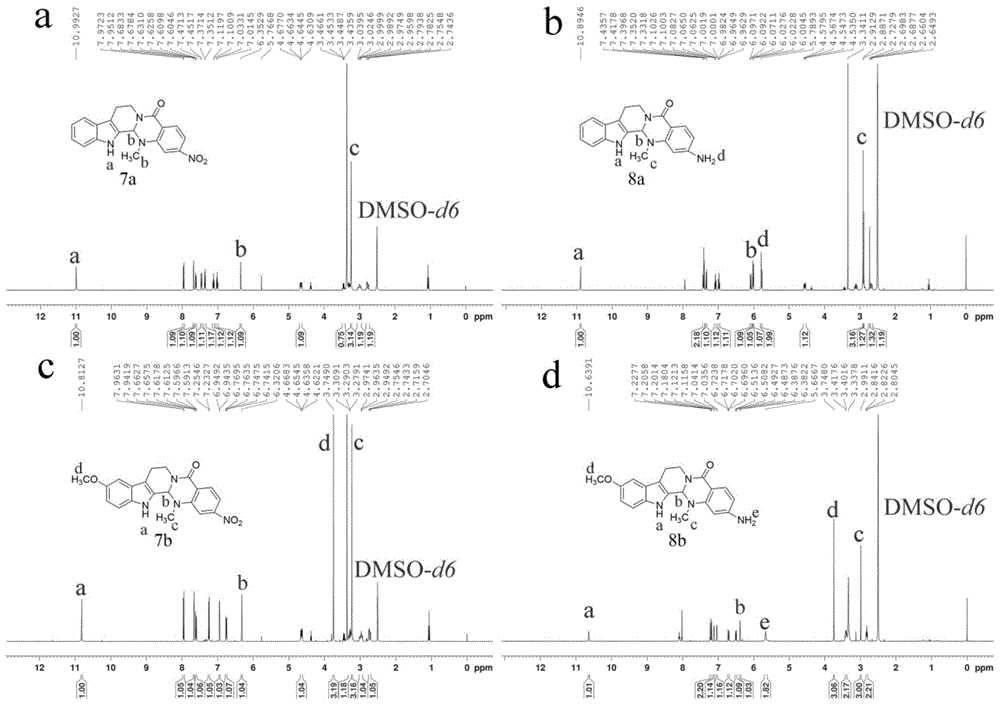
图1为(a)2-no2-evo的核磁共振氢谱,(b)2-nh2-evo的核磁共振氢谱,(c)10-och3-2-no2-evo的核磁共振氢谱,(d)10-och3-2-nh2-evo的核磁共振氢谱;
图2为(a)2-no2-evo和2-nh2-evo的红外光谱,(b)10-och3-2-no2-evo和10-och3-2-nh2-evo的红外光谱;
图3为(a)2-no2-evo和2-nh2-evo的紫外光谱,(b)10-och3-2-no2-evo和10-och3-2-nh2-evo的紫外光谱;
图4为不同浓度的5种化合物与不同细胞培养48h后的存活率。(a)化合物与乳腺癌mda-mb-231细胞共培养48h后的存活率;(b)化合物与结肠癌sw620细胞共培养48h后的存活率;(c)化合物与正常肝lo2细胞共培养48h后的存活率;
图5为(a)不加药组的细胞凋亡,(b)加药组的细胞凋亡。
具体实施方式
本发明通过使用甲氧基取代基的色胺和硝基取代的邻氨基苯甲酸为原料,合成甲氧基修饰的3,4-二氢-β-咔啉和n-甲基-7-硝基靛红酸酐,然后通过缩合反应,得到2-硝基吴茱萸碱(7a)和10-甲氧基-2-硝基吴茱萸碱(7b),然后选择盐酸/氯化亚锡这种温和的方法来进行还原,选择dmf作为溶剂进行反应生成2-氨基吴茱萸碱(8a)和10-甲氧基-2-氨基吴茱萸碱(8b)。用cck-8法进行细胞毒性实验,评价这几种化合物对乳腺癌mda-mb-231细胞和结肠癌sw620细胞以及正常肝lo2细胞的抗增殖作用。用fitc/pi双染进行细胞凋亡的检测。
首先合成2个带有硝基的化合物2-硝基吴茱萸碱(7a)和10-甲氧基-2-硝基吴茱萸碱(7b);然后再通过还原反应生成2-氨基吴茱萸碱(8a)和10-甲氧基-2-氨基吴茱萸碱(8b)。合成路线如下所示:
具体的,将色胺(化合物1a-b)与甲酸乙酯加热回流反应后旋蒸,得油状液体(化合物2a-b);然后加入二氯甲烷溶解,再于冰盐浴条件下,滴入pocl3,反应后旋蒸,得黑褐色油状液体,加入乙酸水溶液后用二氯甲烷萃取,收集的上层液体中加氨水调节ph至9~10,析出黄色固体,抽滤,得浅黄色固体,再用乙酸乙酯萃取滤液并收集上层有机层;将浅黄色固体溶于乙酸乙酯中,合并有机层并旋蒸得黄色固体咔啉化合物(化合物3a、化合物3b)。
将2-氨基-4-硝基苯甲酸、三光气在四氢呋喃中回流反应后冷却至室温,然后将反应液倒入冰水中,然后抽滤,滤饼洗涤、烘干得淡黄色固体7-硝基靛红酸酐(化合物5);然后将7-硝基靛红酸酐与dmf混合,再加入氢化钠,然后滴加碘甲烷,冰水浴下反应后倒入冰水中,然后抽滤,滤饼洗涤、烘干得黄色固体n-甲基-7-硝基靛红酸酐(化合物6)。
将咔啉化合物与n-甲基-7-硝基靛红酸酐在二氯甲烷中回流反应,得橙黄色固体2-硝基吴茱萸碱衍生物(化合物7a、化合物7b);然后将氯化亚锡二水合物、溶剂、盐酸加入2-硝基吴茱萸碱衍生物中,室温~60℃反应15~25小时,然后用naoh调碱性至ph7~9,在用乙酸乙酯萃取,萃取液经过旋蒸蒸干溶剂,残留物用硅胶柱层析,得到氨基吴茱萸碱衍生物(化合物8a、化合物8b)。
实施例一
化合物3,4-二氢-β-咔啉(3a)的合成
在250ml单口烧瓶中加入色胺(1a)4g(m=160.22,24.97mmol),甲酸乙酯120ml(既是反应物又是溶剂),澄清浅黄色液体,加热回流,60℃回流18小时。旋蒸,得油状液体。在烧瓶中加入二氯甲烷至溶解,0-5℃冰盐浴条件下,用滴液漏斗滴入(10分钟滴完)10mlpocl3,冰盐浴条件下反应2h后室温反应3h。旋蒸,得黑褐色油状液体。加入10%乙酸水溶液至全部溶解。用二氯甲烷萃取多次,收集上层(水层)液体。加氨水调节ph至9.5,析出黄色固体,抽滤,得浅黄色固体,用乙酸乙酯萃取滤液并收集上层有机层,将浅黄色固体溶于乙酸乙酯中,合并有机层并旋蒸得黄色固体3,4-二氢-β-咔啉(3a)3.2g。
化合物n-甲基-7-硝基靛红酸酐(6)的合成
在250ml蒸馏烧瓶中,加入2-氨基-4-硝基苯甲酸(4)4.500g(m=182.14,24.7mmol),三光气3.014g(m=296.73,10.16mmol),100ml四氢呋喃。固体全部溶解,颜色变为浅黄色。70℃回流5h,有固体析出,冷却至室温,将反应液倒入300ml冰水中,析出大量沉淀。抽滤,超纯水淋洗3次,二氯甲烷淋洗3次,烘干得淡黄色固体7-硝基靛红酸酐(5)5.01g。在250ml圆底烧瓶中,加入1.015g化合物5(m=208.13,4.88mmol),50mldmf,溶液为黄色液体,加入氢化钠0.300g(m=24,12.5mmol),为红色液体,活化2h。滴加(10分钟滴完)碘甲烷液体3.142g(m=141.94,22.14mmol),冰水浴下反应4h。将反应液倒入300ml冰水中,析出沉淀。抽滤,用超纯水淋洗三次,烘干得黄色固体n-甲基-7-硝基靛红酸酐(6)0.670g。
2-氨基吴茱萸碱(8a)的合成
在250ml圆底烧瓶中,加入1.11g化合物6(m=222.16,5mmol),0.85g化合物3a(m=170.22,5mmol)和100ml二氯甲烷,溶解,呈澄清黄色液体。45℃加热回流18小时。抽滤,得橙黄色固体2-硝基吴茱萸碱(7a,称为2-no2-evo)1.164g。在250ml烧瓶里加入1.164g化合物7a(m=348,3.34mmol),然后依次加入4.00g(m=225.65,17.7mmol)的氯化亚锡二水合物,50mldmf和10ml浓盐酸。溶液为橙红色,之后变为黄色液体。反应过夜,有沉淀析出。用naoh调碱性至ph7-9,用乙酸乙酯萃取,旋蒸,蒸干溶剂,残留物用硅胶柱层析(二氯甲烷:乙醇=80:1)得到黄色固体2-氨基吴茱萸碱(8a,称为2-nh2-evo)100mg。
10-甲氧基-2-氨基吴茱萸碱(8b,称为10-och3-2-nh2-evo)的合成同上述方法,将色胺(1a)替换为色胺(1b),即r1为甲氧基,其余不变。化合物7b称为10-och3-2-no2-evo。
结构表征
(1)核磁共振(1h-nmr)
称取10mg的化合物溶于0.5ml氘代二甲基亚砜(d6-dmso),以四甲基硅烷(tms)作内标,在25℃温度、400mhz频率下进行测试。
(2)红外光谱(ft-ir)
取适量产物粉末置于atr附件上,进行红外光谱的测试。
(3)高分辨质谱
取1mg的化合物用1ml色谱甲醇溶解,冷冻离心,取上清液,稀释最终浓度为5ppm左右,再离心,取800ul上清液加入至进样waters原装小瓶中,在高分辨质谱仪上进行测试。
(4)紫外光谱
将产物溶于乙醇中,配置成10-5m的溶液,用两通石英比色皿在紫外-可见分光光度计上进行测试,扫描波长200-500nm。
细胞培养
(1)细胞复苏
从-78℃冰箱中取出细胞的冻存管在37℃水浴中,使其快速融化。于800r离心5min后;弃去原液,加入含10%胎牛血清的dmem培养基,吹打,制成重悬液,再倒入含有5ml培养液的培养瓶中,摇匀,放于37℃,5%co2培养箱中培养。
(2)细胞换液
细胞培养第二天时,弃掉培养液,用pbs洗一遍,加入5ml新鲜的培养基。
(3)细胞传代(一传三)
当培养瓶内细胞的密度达到80~90%时则需将其传代。首先,将培养液弃掉,用2mlpbs冲洗2遍,加入1ml胰酶,放入37℃培养箱2min,显微镜下看到团聚的细胞正在一点点消化分开变圆,加入1ml培养液终止消化,轻轻吹打下贴壁细胞,转移至15ml离心管内,于800r离心5min后;弃去上清液,再加入1mldmem培养基混匀,各取300ul细胞悬液于3个培养瓶中,摇匀,37℃,5%co2培养箱中培养。传至第四代后方可用于体外细胞实验。
(4)细胞冻存
将一瓶细胞消化离心后,弃去培养液,加入冻存液(90%血清+10%dmso),每个冻存管里500μl冻存液,放在冻存盒-78低温冻存。
体外细胞毒性
(1)细胞种板
分别取处于对数生长期的乳腺癌、结肠癌、正常肝细胞等,将细胞用胰酶进行消化后离心,弃去上清液,加入新鲜培养基制成细胞悬液,经细胞计数板进行细胞的计数,计数后将细胞接种至96孔的培养皿中。接种数量为:每孔为1×104个细胞,100μl细胞悬液。37℃,5%co2培养箱中培养。
(2)加药
孵育24h细胞贴壁贴壁,然后进行加药。5组样品,每组均设置9个浓度即:0.0152,0.0457,0.137,0.412,1.235,3.704,11.11,33.33和100μm。同时设置空白对照,将96孔板中的培养基弃去,加入含有不同药物浓度的100μl的培养基,培养48h。
(3)加cck-8
在孵育48h后,每孔加入10μl的cck-8溶液,再次放入co2的恒温培养箱中孵育4h。利用酶标仪(450nm)测定每孔的od值,计算出各组样品的存活率。
细胞凋亡
取处于对数生长期的乳腺癌mda-mb-231细胞,将细胞用胰酶进行消化后离心,弃去上清液,加入新鲜培养基制成细胞悬液,计数后将细胞接种至24孔的培养皿中。接种数量为:每孔50万个细胞,37℃,5%co2培养箱中孵育24h使得细胞贴壁,然后进行加药。一组空白对照,另一组为浓度20um的2-nh2-evo,各三个孔。孵育48后进行fitc/pe双染,用流式细胞仪测试。
实验结果
化合物的结构表征
(1)1hnmr核磁氢谱及高分辨质谱表征
化合物2-no2-evo、10-och3-2-no2-evo、2-nh2-evo、10-och3-2-nh2-evo的核磁共振氢谱见图1。
其中,图1a为2-no2-evo的氢谱,1hnmr(dmso-d6,400mhz)δ:10.99(s,1h),7.96(d,j=8.4hz,1h),7.68(d,j=2.0hz,1h),7.62(dd,j=8.5,2.1hz,1h),7.46(d,j=7.8hz,1h),7.36(d,j=8.1hz,1h),7.12(t,j=8.0hz,1h),7.01(t,j=7.2hz,1h),6.35(s,1h),4.65(dd,j=13.0,5.4hz,1h),3.44~3.47(m,1h),3.24(s,3h),2.95~3.04(m,1h),2.74~2.79(m,1h)。化学位移10.99ppm的单峰归属于吲哚环上-nh-质子(a),6.35ppm的单峰归属于-ch-质子(b),3.24ppm的单峰归属于的甲基-n-ch3(c)。
图1b为2-nh2-evo的氢谱,1hnmr(dmso-d6,400mhz)δ:10.89(s,1h),7.42(t,j=7.8hz,2h),7.34(d,j=8.1hz,1h),7.06~7.10(m,1h),6.96~7.00(m,1h),6.07~6.10(m,1h),6.00~6.02(m,1h),6.00(s,1h),5.79(s,2h),4.56(dd,j=13.0,4.9hz,1h),2.91(s,3h),2.89(s,1h),2.73(s,1h),2.64~2.66(m,1h).ms(esi):319.15484(c19h18n4o,[m+h])。化学位移10.89ppm的单峰归属于吲哚环上-nh-质子(a),6.00ppm的单峰归属于-ch-质子(b),2.91ppm的单峰归属于-n-ch3(c)的甲基,5.79归属于-nh2质子(d)。高分辨质谱测得质荷比为319.15484[m+h],理论值为318[m]。
图1c为10-och3-2-no2-evo的氢谱,1hnmr(dmso-d6,400mhz)δ:10.81(s,1h),7.95(d,j=8.5hz,1h),7.66(d,j=2.1hz,1h),7.60(dd,j=8.5,2.1hz,1h),7.24(d,j=8.8hz,1h),6.95(d,j=2.3hz,1h),6.76(dd,j=8.8,2.4hz,1h),6.32(s,1h),4.65(dd,j=13.0,5.5hz,1h),3.75(s,3h),3.29~3.28(m,1h),3.23(s,3h),2.97~2.95(m,1h),2.75~2.71(m,1h)。化学位移10.81ppm的单峰归属于吲哚环上-nh-质子(a),6.32ppm的单峰归属于-ch-质子(b),3.23ppm的单峰归属于-n-ch3(c)的甲基,3.75ppm的单峰归属于-och3(d)的甲基。
图1d为10-och3-2-nh2-evo的氢谱,1hnmr(dmso-d6,400mhz)δ:10.64(s,1h),7.18~7.22(m,2h),7.12(d,j=2.2hz,1h),7.04(d,j=2.3hz,1h),6.71(dd,j=8.7,2.4hz,1h),6.50(dd,j=8.4,2.2hz,1h),6.39(d,j=2.2hz,1h),5.67(s,2h),3.74(s,3h),3.40~3.42(m,2h),2.99(s,3h),2.82(t,j=7.4hz,2h).ms(esi):349.16529(c20h18n3o2,[m+h])。化学位移为10.64ppm的单峰归属于吲哚环上-nh-质子(a),6.38ppm的单峰归属于-ch-质子(b),2.99ppm的单峰归属于-n-ch3(c)的甲基,3.74ppm的单峰归属于-och3(d)的甲基,5.67ppm的单峰归属于-nh2质子(e)。高分辨质谱测得质荷比为349.16529[m+h],理论值为348[m]。
(2)红外光谱
化合物2-no2-evo、2-nh2-evo、10-och3-2-no2-evo、10-och3-2-nh2-evo的红外光谱图如图2。图2a为2-no2-evo和2-nh2-evo的红外吸收光谱图,2-no2-evo在波数3353cm-1处有一个尖峰,这是分子中吲哚环上亚氨基-nh-的特征吸收峰。硝基还原成氨基后,出现氨基的伸缩振动新峰,因此,2-nh2-evo除了在3356cm-1处的亚氨基吸收峰外,还在3260cm-1和3216cm-1处出现一个双峰,这是氨基-nh2的特征吸收峰。同理,图2b为10-och3-2-no2-evo和10-och3-2-nh2-evo的红外吸收光谱图,10-och3-2-no2-evo在3402cm-1处的尖峰为吲哚环上亚氨基-nh-的特征吸收峰。10-och3-2-nh2-evo除了在3446cm-1处的亚氨基吸收峰外,还在3340cm-1和3302cm-1处出现一个双峰,此峰为侧壁上的氨基的特征吸收峰。
(3)紫外光谱
化合物2-no2-evo、10-och3-2-no2-evo、2-nh2-evo、10-och3-2-nh2-evo的紫外光谱如图3。图3a为2-no2-evo和2-nh2-evo的紫外吸收光谱图,化合物2-no2-evo在251nm处有最大吸收,2-nh2-evo在292nm处有最大吸收。图3b为10-och3-2-no2-evo和10-och3-2-nh2-evo的紫外吸收光谱图,10-och3-2-no2-evo在249nm处有最大吸收,10-och3-2-nh2-evo在275nm处有最大吸收。结果表明硝基还原成氨基后,紫外吸收光谱红移,因为氨基为供电基团,硝基为吸电子基团,修饰上氨基后,苯环负电荷密度变大,使得紫外波长红移。
综上所述,核磁共振氢谱,高分辨质谱,红外光谱的测试结果表明,已成功合成了2-nh2-evo和10-och3-2-nh2-evo这两种化合物,并通过紫外吸收光谱研究了化合物在紫外区的吸收曲线。
体外细胞毒性评价结果
为了评价修饰后的吴茱萸碱衍生物的体外细胞毒性,以evo为对照,将4种化合物2-no2-evo、2-nh2-evo、10-och3-2-no2-evo、10-och3-2-nh2-evo与两种癌细胞乳腺癌细胞mda-mb-231、结肠癌细胞sw620和一种正常肝细胞lo2共同孵育48小时后,用cck-8试剂盒进行毒性结果的评定,并计算存活率,结果如图4所示,2.5a和2.5b表明,化合物对乳腺癌细胞mda-mb-231和结肠癌细胞sw620抑制趋势一致,其中evo、2-no2-evo、2-nh2-evo、10-och3-2-no2-evo这四种化合物随着药物浓度的增大,细胞存活率降低,呈现出剂量-浓度依赖,但是化合物10-och3-2-nh2-evo随着药物浓度的增大,细胞存活率几乎不变,对肿瘤细胞几乎没有抑制作用。图2.5c表明这5种化合物对正常肝细胞lo2的毒性很小。
为了进一步比较化合物的活性,根据存活率与浓度的量效关系曲线,计算了五种化合物对两种癌细胞的存活率,结果见表1、表2,统计学分析采用spss22统计软件,采用单因素方差分析比较各药物组与对照组间有无统计学差异,以p<0.05为具有统计学意义。
注:*表示同一化合物,与不加药组相比,在某一浓度下的细胞存活率具有统计学差异,p<0.05。®表示在某一浓度下,与evo相比,2-nh2-evo作用后细胞存活率具有统计学差异,p<0.05。
evo、2-no2-evo、2-nh2-evo及10-och3-2-no2-evo对乳腺癌细胞的ic50值分别1.034μm,3.869μm,0.79μm及44.95μm,10-och3-2-nh2-evo的ic50远大于100μm。ic50值越小,表明,活性越高。与evo相比,2-nh2-evo的ic50值较小,活性较好,而2-no2-evo、10-och3-2-no2-evo及10-och3-2-nh2-evo的活性降低。同理,evo和2-nh2-evo对结肠癌细胞的ic50分别为3.473um和1.28um,而2-no2-evo、10-och3-2-no2-evo及10-och3-2-nh2-evo的ic50均大于100um,表明2-nh2-evo对结肠癌细胞具有很好的抑制作用。
综上所述,在evo侧壁2位上修饰硝基会使活性降低,但是硝基还原成氨基后,活性增加;在10位修饰甲氧基后,同时在2位修饰硝基或氨基后,活性均降低。
细胞凋亡结果
为了进一步研究化合物对乳腺癌细胞的抗增殖作用机制,将乳腺癌细胞与20um的2-nh2-evo溶液共同孵育48h,用annexinv-egfp/pi双染法检测细胞凋亡状况,并用流式细胞仪进行结果的测定,如图5。其中5a为空白对照不加药组,5b为加药组(20um,2-nh2-evo),空白组中95.12%的细胞处于正常状态,2.76%的细胞处于早凋状态,1.65%的细胞处于晚凋状态,0.47%的细胞处于死亡状态。给药组中,41.14%的细胞处于正常状态,8.39%的细胞处于早凋状态,48.54%的细胞处于晚凋状态,1.93%的细胞处于死亡状态。不加药组总的凋亡率为4.41%,加药组总的凋亡率为56.93%,结果表明2-nh2-evo能够显著抑制乳腺癌细胞的凋亡。
本发明通过对吴茱萸碱进行结构修饰,研究了修饰的吴茱萸碱衍生物的抗肿瘤活性,与先导化合物吴茱萸碱相比,本发明的化合物2-nh2-evo对乳腺癌mda-mb-231细胞表现出最好的抗肿瘤活性,对正常肝细胞的毒性较小,能够显著诱导乳腺癌细胞的凋亡,有望发展成为有潜力的抗肿瘤小分子化合物。
- 还没有人留言评论。精彩留言会获得点赞!