一种从发酵液中分离提取原儿茶酸的方法与流程
本发明属于生化分离技术领域,具体涉及一种从发酵液中分离提取原儿茶酸的方法。
背景技术:
原儿茶酸又名3,4-二羟基苯甲酸,是天然存在于多种植物中的一种水溶性酚酸类物质,并且是很多中药的活性物质,具有祛痰平喘、抗菌、镇痛、抗凝血、抗氧化、抗肿瘤和神经保护等多种作用,有较高的医药价值。原儿茶酸的分子结构中含有两个酚羟基和一个羧基,可以进行分子间酯化反应,形成高聚物,制备高分子材料,有广泛的应用价值。
目前,原儿茶酸的生产方法有化学合成和天然植物提取法。化学合成反应需高温、强酸(碱)条件,危险系数高,环境危害大,对设备要求高。天然植物提取法需大量植物,为不可再生资源,且植物成分复杂,目标产物一般含量较低。例如:一种利用龙眼壳制备原儿茶酸的工艺(专利申请号201710596858.2)中以龙眼壳为原料,其原儿茶酸含量较低,制备过程中会产生大量固体废弃物;番石榴叶中9种抗氧化多酚的提取纯化方法(专利申请号201710113566.9)中以番石榴叶为原料提取原儿茶酸,其成分复杂,很难获得纯度较高原儿茶酸,只能获得多种产物的复合物;一种用稀碱溶液提取-固相萃取柱纯化原儿茶酸的方法与流程(专利申请号201711044028.5)中以四季青叶为原料,提取过程中需要使用稀碱、乙酸乙酯、乙醚等诸多诸多化学溶剂,并且还需要用到成本较高固相萃取柱萃取分离原料中的杂质,成本高、效率低,环境危害大。
微生物有生长繁殖快,生长周期短,产量高,杂质少,可以进行大规模生产的特点,可以克服化学法合成的不足之处,非常适合用于工业化生产;其培养方法简单,原料来源丰富,价格低廉,经济效益高。微生物发酵生产原儿茶酸素最大的特点是产物专一性高、结构类似的有机杂质相对较少。
技术实现要素:
本发明的目的是从发酵液中分离提取原儿茶酸。
本发明首先保护一种从发酵液中分离提取原儿茶酸的方法,依次包括如下步骤:
(1)取含有原儿茶酸的发酵液,除去菌体,收集澄清发酵液;
(2)调节所述澄清发酵液ph值至3.0-4.0(如3.0-3.5、3.5-4.0、3.0、3.5或4.0),得到酸化液;
(3)将所述酸化液静置1-3h(如1-2h、2-3h、1h、2h或3h)或75-85℃(如75-80℃、80-85℃、75℃、80℃或85℃)搅拌20-40min(如20-30min、30-40min、20min、30min或40min),除去不溶物,得酸化澄清液;
(4)用乙酸乙酯萃取所述酸化澄清液2-4次(如2次、3次或4次),合并乙酸乙酯相,得到有机相;
(5)将所述有机相进行干燥,获得固体沉淀;用水溶解固体沉淀,得到原儿茶酸粗提液;
(6)将所述原儿茶酸粗提液进行脱色,结晶,得到原儿茶酸。
上述方法中,所述步骤(1)中,除去菌体的方式可为过滤或离心。离心可为管式离心机离心或碟片式离心机离心。离心速度不低于8000rpm。过滤可为陶瓷膜过滤或中空纤维膜过滤。具体的,过滤选用膜孔径为100nm的陶瓷膜过滤,操作压力为0.3mpa。具体的,过滤选用膜孔径为0.22μm的中空纤维膜过滤。
上述方法中,所述步骤(2)中,调节澄清发酵液ph值可通过加入盐酸或硫酸实现。调节ph值具体可为加入浓度为6mol/l盐酸,调节ph值至3.0-4.0(如3.0-3.5、3.5-4.0、3.0、3.5或4.0)。调节ph值具体可为加入浓硫酸,调节ph值至3.0-4.0(如3.0-3.5、3.5-4.0、3.0、3.5或4.0)。
上述方法中,所述步骤(3)中,除去不溶物的方式可为过滤或离心。离心可为管式离心机离心或碟片式离心机离心。离心速度不低于8000rpm。过滤可为陶瓷膜过滤或中空纤维膜过滤。具体的,过滤选用膜孔径为100nm的陶瓷膜过滤,操作压力为0.3mpa。具体的,过滤选用膜孔径为0.22μm的中空纤维膜过滤。
上述方法中,所述步骤(4)中,每次萃取的时间可为1-3h(如1-2h、2-3h、1h、2h或3h)。用乙酸乙酯萃取酸化澄清液时,酸化澄清液和乙酸乙酯的体积比可为1:1。
上述方法中,所述步骤(5)中,干燥的温度可为50-80℃(如50-60℃、60-70℃、70-80℃、50℃、60℃、70℃或80℃)。所述干燥可为减压浓缩干燥。
上述方法中,所述步骤(5)中,用水溶解固体沉淀可为用50℃-80℃(如50-60℃、60-70℃、70-80℃、50℃、60℃、70℃或80℃)的去离子水溶解固体沉淀。
上述方法中,所述步骤(6)中,所述脱色通过加入活性炭实现。
上述方法中,所述步骤(6)具体可为:向原儿茶酸粗提液中加入活性炭脱色,结晶,得到晶体;将晶体纯化2次以上(如2次、3次、4次),得到原儿茶酸;
每次纯化步骤为:向晶体纯水溶液中加入活性炭脱色,结晶。
上述任一所述活性炭的使用量为0.5%(m/v)以上(如0.5%(m/v)、1.0%(m/v)、)。
上述任一所述结晶的温度为4℃以下(如4℃)。
上述任一所述含有原儿茶酸的发酵液是由生产原儿茶酸的菌株发酵培养获得的。所述生产原儿茶酸的菌株具体可为大肠杆菌ma03-50。
上述任一所述含有原儿茶酸的发酵液具体可为大肠杆菌ma03-50发酵液。大肠杆菌ma03-50发酵液的制备方法可如下:将大肠杆菌ma03-50单菌落接种至3ml一级种子培养基,30℃、250rpm培养16h,得到一级种子培养液;将2ml一级种子培养液接种于200ml二级种子培养基,37℃、250rpm培养24h,得到二级种子培养液;将200ml二级种子培养液接种于2l初始发酵罐培养基,37℃,ph6.5(通过浓氨水调控ph),溶氧20%的条件下发酵。发酵开始后,待发酵罐中葡萄糖浓度降低到1g/l以下时,开始用浓度为500g/l的葡萄糖溶液开始补料,控制补料速度使发酵罐中葡萄糖浓度小于1g/l。上述制备方法、大肠杆菌ma03-50、一级种子培养基、二级种子培养基和初始发酵罐培养基均记载于如下中国发明专利文献cn109943512a中。
实验证明,采用本发明提供的方法从发酵液中分离提取原儿茶酸,纯度可达98%以上,收率可达80%以上。该方法分离纯化过程简单,产品纯度高,成本低,适合连续生产,符合工业化要求。本发明具有重要的应用价值。
附图说明
图1为实施例1中步骤三原儿茶酸粗提液甲的液相色谱图。
图2为实施例1中步骤三溶液1的液相色谱图。
图3为实施例1中步骤三母液的液相色谱图。
图4为实施例1中步骤三溶液2的液相色谱图。
图5为实施例1中步骤三溶液3的液相色谱图。
图6为实施例2晶体3水溶液的液相色谱图。
图7为实施例3晶体c水溶液的液相色谱图。
图8为实施例4晶体d水溶液的液相色谱图。
具体实施方式
下面结合具体实施方式对本发明进行进一步的详细描述,给出的实施例仅为了阐明本发明,而不是为了限制本发明的范围。以下提供的实施例可作为本技术领域普通技术人员进行进一步改进的指南,并不以任何方式构成对本发明的限制。
下述实施例中的实验方法,如无特殊说明,均为常规方法,按照本领域内的文献所描述的技术或条件或者按照产品说明书进行。下述实施例中所用的材料、试剂等,如无特殊说明,均可从商业途径得到。
下述实施例中,大肠杆菌ma03-50发酵液的制备方法如下:将大肠杆菌ma03-50单菌落接种至3ml一级种子培养基,30℃、250rpm培养16h,得到一级种子培养液;将2ml一级种子培养液接种于200ml二级种子培养基,37℃、250rpm培养24h,得到二级种子培养液;将200ml二级种子培养液接种于2l初始发酵罐培养基,37℃,ph6.5(通过浓氨水调控ph),溶氧20%的条件下发酵。发酵开始后,待发酵罐中葡萄糖浓度降低到1g/l以下时,开始用浓度为500g/l的葡萄糖溶液开始补料,控制补料速度使发酵罐中葡萄糖浓度小于1g/l。大肠杆菌ma03-50已于2017年10月25日保藏于中国微生物菌种保藏管理委员会普通微生物中心(简称cgmcc,地址:北京市朝阳区北辰西路1号院3号,中国科学院微生物研究所,邮编100101),保藏号为cgmccno.14834。上述制备方法、大肠杆菌ma03-50、一级种子培养基、二级种子培养基和初始发酵罐培养基均记载于如下中国发明专利文献cn109943512a中。
下述实施例中,收率=晶体质量/澄清发酵液中原儿茶酸含量×100%。
实施例1、从发酵液中分离提取原儿茶酸的方法的建立
一、最佳萃取溶剂的获得
原儿茶酸在水、乙醇和乙酸乙酯三种溶剂中不同温度下的溶解度对于发酵液中原儿茶酸分离、纯化、结晶工艺开发非常重要,目前无法从现有技术中找到完整物理化学数据。
按照常规溶解度测定的方法,测定原儿茶酸在水、乙醇和乙酸乙酯三种溶剂中不同温度下的溶解度。
测定结果见表1。结果表明,原儿茶酸在乙酸乙酯的溶解度是水的10倍以上。因此,常温下发酵液调酸(把原儿茶酸从离子形态转变为分子形态)后加入乙酸乙酯进行萃取,可以快速把原儿茶酸全部萃取到有机相中。此外,发酵液产原儿茶酸的有机杂质少,但是发酵液中还是存在大量的发酵原料残留(如酵母膏胶体、无机盐、大分子物质等),通过有机溶剂乙酸乙酯萃取可以完美转移原儿茶酸而原来的发酵残留(几乎都是水溶性成分)留在水溶液中,实现水溶性杂质的快速分离。
表1.原儿茶酸在不同温度与溶剂中溶解度的测定结果
上述结果表明,有机溶剂乙酸乙酯为最佳的萃取溶剂。
二、从发酵液中分离提取原儿茶酸的方法的建立
本发明的发明人经过大量实验,建立了从发酵液中分离提取原儿茶酸的方法。具体步骤如下:
1、取发酵液(含有原儿茶酸),除去菌体,收集澄清发酵液。
除去菌体的方式为过滤。过滤具体选用膜孔径为100nm的陶瓷膜过滤,操作压力为0.3mpa。
2、完成步骤1后,取澄清发酵液,调节ph值至4.0以下,得到酸化液。
调节ph值至4.0以下具体为加入浓度为6mol/l盐酸,调节ph值至3.5。
3、完成步骤2后,取酸化液,室温静置2h,除去不溶物,得酸化澄清液。
除去不溶物的方式为过滤。过滤具体选用膜孔径为100nm的陶瓷膜过滤,操作压力为0.3mpa。
4、完成步骤3后,取酸化澄清液,用等体积乙酸乙酯萃取3次,合并乙酸乙酯相,得到有机相。
每次萃取的时间为2h。
5、完成步骤4后,取有机相,60℃减压浓缩干燥,得到固体沉淀;取固体沉淀,加入去离子水(具体可为温度为50℃-80℃的去离子水)复溶,得到原儿茶酸粗提液。
6、完成步骤5后,取原儿茶酸粗提液,加入活性炭脱色,过滤(目的为去除活性炭),降温结晶,得到原儿茶酸。
活性炭的添加量不低于0.5%(m/v)。
结晶的温度不高于4℃。
三、从发酵液中分离提取原儿茶酸的方法的优化
1、当发酵液为大肠杆菌ma03-50发酵液时,按照上述步骤1-5,得到原儿茶酸粗提液甲。
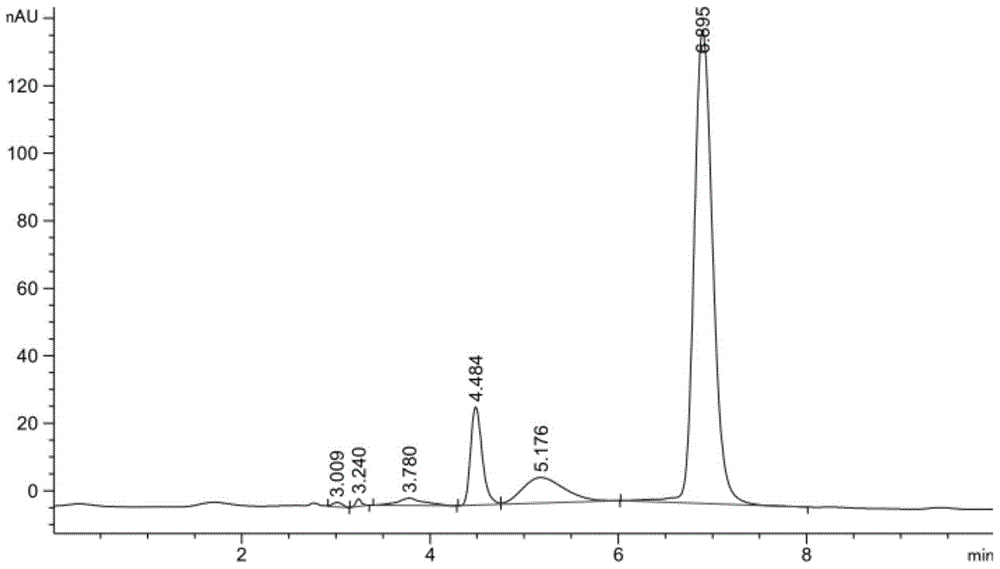
将原儿茶酸粗提液甲进行液相色谱检测,结果见图1(保留时间6.895min为原儿茶酸,保留时间4.484min为没食子酸)。结果表明,原儿茶酸粗提液甲中主要含有原儿茶酸,还有没食子酸和一些杂质。
2、取原儿茶酸粗提液甲,加入活性炭脱色(活性炭的添加量不低于0.5%(m/v)),过滤(目的为去除活性炭),4℃结晶,得到晶体1和母液。
将晶体1用去离子水(具体可为温度为50℃-80℃的去离子水)溶解,得到溶液1。
将溶液1和母液分别进行液相色谱检测,结果依次见图2(保留时间6.928min为原儿茶酸,保留时间4.499为没食子酸)和图3(保留时间6.818min为原儿茶酸,保留时间4.465min为没食子酸)。结果表明,溶液1中主要杂质没食子酸已经大幅度减少,没食子酸主要在母液中。
3、取溶液1,加入活性炭脱色(活性炭的添加量不低于0.5%(m/v)),过滤(目的为去除活性炭),4℃结晶,得到晶体2。
将晶体2用去离子水(具体可为温度为50℃-80℃的去离子水)溶解,得到溶液2。
将溶液2进行液相色谱检测,结果见图4(保留时间6.876为原儿茶酸)。结果表明,溶液2中原儿茶酸的纯度为96%。
4、取溶液2,加入活性炭脱色(活性炭的添加量不低于0.5%(m/v)),过滤(目的为去除活性炭),4℃结晶,得到晶体3。
将晶体3用去离子水(具体可为温度为50℃-80℃的去离子水)溶解,得到溶液3。
将溶液3进行液相色谱检测,结果见图5(保留时间7.079为原儿茶酸)。结果表明,溶液3中原儿茶酸的纯度为98%。
由此可见,乙酸乙酯萃取液蒸干并在纯水中进行重结晶三次,可以快速去除主要杂质得到高纯度的原儿茶酸晶体。
因此,为了获得纯度较高的原儿茶酸,步骤二中建立的方法的步骤6优化为:完成步骤5后,取原儿茶酸粗提液,加入活性炭脱色,降温,得到晶体;将晶体纯化2次以上,得到原儿茶酸;纯化方法为:向晶体纯水溶液中加入活性炭脱色,降温。活性炭的添加量不低于0.5%(m/v)。结晶的温度不高于4℃。
步骤二中1,除去菌体的方式为离心或过滤。离心为管式离心机离心或碟片式离心机离心。离心速度不低于8000rpm。过滤为陶瓷膜过滤或中空纤维膜过滤。
步骤二中2,调节ph值一般加入盐酸或硫酸。调节ph值具体为加入浓度为6mol/l盐酸,调节ph值至3.0-4.0。
步骤二中3,室温静置2h可替换为80℃搅拌混匀30min。
步骤二中3,除去不溶物的方式为离心或过滤。离心速度不低于8000rpm。离心为管式离心机离心或碟片式离心机离心。过滤为陶瓷膜过滤或中空纤维膜过滤。
步骤二中4,每次萃取的时间为1-2h。
步骤二中5,干燥的温度为50-80℃(如50-60℃、60-80℃、50℃、60℃或80℃)。
实施例2、从大肠杆菌ma03-50发酵液中分离提取原儿茶酸的实验一
1、取大肠杆菌ma03-50发酵液,除去菌体,收集澄清发酵液1.0l。
澄清发酵液中原儿茶酸的浓度为66g/l。
除去菌体的方式为过滤。过滤具体选用膜孔径为0.22μm的中空纤维膜过滤。
2、完成步骤1后,取澄清发酵液,调节ph值至4.0以下,得到酸化液。
调节ph值至4.0以下具体为加入浓硫酸,调节ph值至3.5。
3、完成步骤2后,取酸化液,80℃搅拌混匀30min,除去不溶物,得酸化澄清液。
除去不溶物的方式为过滤。过滤具体选用膜孔径为0.22μm的中空纤维膜过滤。
4、完成步骤3后,取酸化澄清液,用等体积乙酸乙酯萃取3次,合并乙酸乙酯相,得到3l有机相。
每次萃取的时间为1h。
5、完成步骤4后,取有机相,60℃旋转蒸发、干燥,得到固体沉淀;取固体沉淀,加入1l去离子水(具体可为温度为50℃-80℃的水)复溶,得到原儿茶酸粗提液。
6、完成步骤5后,取原儿茶酸粗提液,加入0.5%活性炭搅拌混匀30min后过滤,减压浓缩至400ml,缓慢冷却到4℃结晶,结晶完成后过滤出晶体1。晶体1溶解于300ml去离子水(具体可为温度为50℃-80℃的水)中,加入0.5%活性炭搅拌混匀30min后过滤,缓慢冷却到4℃结晶,结晶完成后过滤出晶体2。晶体2溶解于250ml去离子水(具体可为温度为50℃-80℃的水)中,加入0.5%活性炭搅拌混匀30min后过滤,缓慢冷却到4℃结晶,结晶完成后过滤出晶体3。
最终晶体3为53.6g,收率为81.0%(53.6g/(66g/l×1.0l)×100%=81.0%)。
将晶体3溶于去离子水进行液相色谱检测,结果见图6。结果表明,原儿茶酸峰面积百分比98.2%,质量浓度百分比98.7%。
实施例3、从大肠杆菌ma03-50发酵液中分离提取原儿茶酸的实验二
1、取大肠杆菌ma03-50发酵液,除去菌体,收集澄清发酵液2.0l。
澄清发酵液中原儿茶酸的浓度为66g/l。
除去菌体的方式为过滤。过滤具体选用膜孔径为100nm的陶瓷膜过滤,操作压力为0.3mpa。
2、完成步骤1后,取澄清发酵液,调节ph值至4.0以下,得到酸化液。
调节ph值至4.0以下具体为加入浓度为6mol/l盐酸,调节ph值至3.2。
3、完成步骤2后,取酸化液,80℃搅拌混匀30min,除去不溶物,得酸化澄清液。
除去不溶物的方式为过滤。过滤具体选用膜孔径为0.22μm的中空纤维膜过滤。
4、完成步骤3后,取酸化澄清液,用等体积乙酸乙酯萃取3次,合并乙酸乙酯相,得到6l有机相。
每次萃取的时间为1h。
5、完成步骤4后,取有机相,60℃旋转蒸发、干燥,得到固体沉淀;取固体沉淀,加入1l去离子水(具体可为温度为50℃-80℃的去离子水)复溶,得到原儿茶酸粗提液。
6、完成步骤5后,取原儿茶酸粗提液,加入0.5%活性炭搅拌混匀30min后过滤,缓慢冷却到4℃结晶,结晶完成后过滤出晶体a。晶体a溶解于800ml去离子水(具体可为温度为50℃-80℃的去离子水)中,加入0.5%活性炭搅拌混匀30min后过滤,缓慢冷却到4℃结晶,结晶完成后过滤出晶体b。晶体b溶解于600ml去离子水(具体可为温度为50℃-80℃的去离子水水)中,加入0.5%活性炭搅拌混匀30min后过滤,缓慢冷却到4℃结晶,结晶完成后过滤出晶体c。
最终晶体c为106.0g,收率为80.0%(106.0/(66g/l×2.0l)×100%=80.0%)。
将晶体c溶于去离子水进行液相色谱检测,结果见图7。结果表明,原儿茶酸峰面积百分比98.7%,质量浓度百分比99.0%。
实施例4、从大肠杆菌ma03-50发酵液中分离提取原儿茶酸的实验三
1、取大肠杆菌ma03-50发酵液,除去菌体,收集澄清发酵液18.0l。
澄清发酵液中原儿茶酸的浓度为66g/l。除去菌体的方式为过滤。过滤具体选用膜孔径为0.22μm的中空纤维膜过滤。
2、完成步骤1后,取澄清发酵液,调节ph值至4.0以下,得到酸化液。
调节ph值至4.0以下具体为加入浓硫酸,调节ph值至3.5。
3、完成步骤2后,取酸化液,室温静置2h,除去不溶物,得酸化澄清液。
除去不溶物的方式为过滤。过滤具体选用膜孔径为0.22μm的中空纤维膜过滤。
4、完成步骤3后,取酸化澄清液,用等体积乙酸乙酯萃取3次,合并乙酸乙酯相,得到60l有机相。
每次萃取的时间为1.5h。
5、完成步骤4后,取有机相,60℃旋转蒸发、干燥,得到固体沉淀;取固体沉淀,加入6.7l去离子水(具体可为温度为50℃-80℃的去离子水)复溶,得到原儿茶酸粗提液。
干燥的温度为80℃。
6、完成步骤5后,取原儿茶酸粗提液,加入1.0%活性炭搅拌混匀30min后过滤,缓慢冷却到4℃结晶,结晶完成后过滤出晶体a。晶体a溶解于4l去离子水(具体可为温度为50℃-80℃的去离子水)中,加入1.0%活性炭搅拌混匀30min后过滤,缓慢冷却到4℃结晶,结晶完成后过滤出晶体b。晶体b溶解于3.5l去离子水(具体可为温度为50℃-80℃的去离子水)中,加入1.0%活性炭搅拌混匀30min后过滤,缓慢冷却到4℃结晶,结晶完成后过滤出晶体c。晶体c溶解于3.0l去离子水(具体可为温度为50℃-80℃的去离子水)中,加入1.0%活性炭搅拌混匀30min后过滤,缓慢冷却到4℃结晶,结晶完成后过滤出晶体d。
最终晶体d为978.0g,收率为82.3%(978.0/(66g/l×18.0l)×100%=82.3%)。
将晶体d溶于去离子水进行液相色谱检测,结果见图8。结果表明,原儿茶酸峰面积百分比96.6%,质量浓度百分比97.5%。
以上对本发明进行了详述。对于本领域技术人员来说,在不脱离本发明的宗旨和范围,以及无需进行不必要的实验情况下,可在等同参数、浓度和条件下,在较宽范围内实施本发明。虽然本发明给出了特殊的实施例,应该理解为,可以对本发明作进一步的改进。总之,按本发明的原理,本申请欲包括任何变更、用途或对本发明的改进,包括脱离了本申请中已公开范围,而用本领域已知的常规技术进行的改变。按以下附带的权利要求的范围,可以进行一些基本特征的应用。
- 还没有人留言评论。精彩留言会获得点赞!