一种依维莫司乙基化杂质的制备方法与流程
1.本发明属于医药化工领域,具体涉及一种依维莫司乙基化杂质的制备方法。
背景技术:
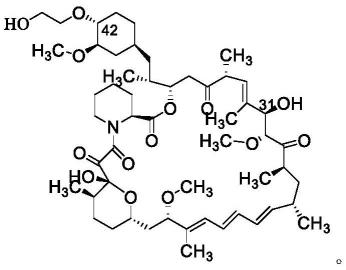
2.依维莫司(everolimus)是诺华(novartis)公司研发的新一代的大环内酯类免疫抑制剂及抗肿瘤药物,其由雷帕霉素的42-oh衍生为42-o-(2-羟乙基)而成,故依维莫司又称42-o-(2-羟乙基)-雷帕霉素。主要适用于舒尼替尼或索拉非尼治疗失败后晚期肾癌细胞癌患者的治疗,结构式如下:
[0003][0004]
目前报道的依维莫司常用的合成方法有如下两种:
[0005]
方法一:在有机碱条件下,以雷帕霉素或者硅基选择性保护雷帕霉素31-位羟基和单保护氧乙基三氟甲磺酸酯为反应物,所得产物脱去硅醚保护即得到产品依维莫司。
[0006]
方法二:以硅基选择性保护雷帕霉素31-位羟基或雷帕霉素衍生物为原料,与三氟甲磺酸酐反应,将原料的42位羟基活化,后与单保护的乙二醇反应,再脱硅醚保护基得到产品依维莫司。
[0007]
对两种合成方法研究发现,依维莫司大环内酯与乙醇接触会产生乙基化杂质,结构如下:
[0008][0009]
由于其结构与依维莫司类似,该杂质的稳定性等物化性质都与产物极其相似,很难通过降解、分离等常规方法大量获得,而目前关于依维莫司乙基化杂质的合成专利文献较少,仅有中国专利申请cn104530112报道了该乙基化杂质合成的制备方法,采用依维莫司和无水乙醇为反应物,在酸性条件下反应得到乙基化杂质,这种方法反应时间过长,并且在
酸性条件下,依维莫司容易产生手性异构互换,导致合成过程中会有异构化杂质产生,与目标产物分离困难,收率过低,工业化成本高。
[0010]
因此,为依维莫司乙基化杂质探究一种生产成本低,操作简便、收率更高的工艺路线仍然是目前需要解决的问题。
技术实现要素:
[0011]
为优化依维莫司乙基化杂质合成工艺,本发明提供了一种新的依维莫司乙基化杂质的制备方法,该方法反应条件温和、路线短、成本低、操作简单,所得乙基化杂质纯度高、收率高。
[0012]
本发明具体通过如下技术方案实现:
[0013][0014]
一种依维莫司乙基化杂质的制备方法,包括如下步骤:室温下将化合物sm-1加入乙醇及有机溶剂中溶解,控温加入无机碱,继续控温搅拌反应得依维莫司乙基化杂质。
[0015]
优选地,所述的无机碱选自氢氧化钠、碳酸钠、碳酸钾中的一种或其组合,其中特别优选氢氧化钠。
[0016]
优选地,所述的化合物sm-1、无机碱的投料摩尔比为1:0.5~1.5,其中特别优选1:0.8。
[0017]
优选地,所述的有机溶剂选自三氯甲烷、1.4-二氧六环、四氢呋喃中的一种或其组合,其中特别优选四氢呋喃。
[0018]
优选地,所述有机溶剂与乙醇的体积比为2:1~4:1。
[0019]
优选地,所述的加入无机碱及加入碱后反应温度为30~65℃,其中特别优选40~45℃
[0020]
在一优选方案中,反应结束后需进行后处理操作,具体的为:反应完毕后向反应液加入去离子水和萃取剂,搅拌分液,水相用萃取剂提取一遍,合并有机相,分别用饱和nahco3溶液、饱和食盐水洗涤分液,有机相用无水硫酸钠干燥后,减压浓缩,柱层析分离(洗脱剂先用v
石油醚
:v
乙酸乙酯
=1:1洗脱前杂,后用乙酸乙酯洗脱);所述萃取溶剂为二氯甲烷、三氯甲烷、乙酸乙酯中的一种或其组合。
[0021]
与现有技术相比,本发明取得的技术效果是:
[0022]
1、本发明提供了一种合成依维莫司乙基化杂质的新方法,该方法可以避免酸性条件导致的手性异构化杂质的产生,更利于目标产物的纯化,同时较原合成方法反应时间缩短,大大降低了人工成本,操作简单,收率、纯度都有很大的提高,更适合工业化生产;
[0023]
2、本发明制备方法操作简单所得杂质纯度高、收率高。
附图说明
[0024]
附图1:化合物i的核磁共振氢谱
[0025]
附图2:化合物i的hplc图谱(实施例4)
[0026]
附图3:化合物i的质谱图谱
具体实施方式
[0027]
下面通过实施例来进一步说明本发明。应该正确理解的是:本发明的实施例仅仅是用于说明本发明,而不是对本发明的限制,所以,在本发明的方法前提下对本发明的简单改进均属本发明要求保护的范围。
[0028]
实施例1
[0029]
室温下,将依维莫司(10.00g,0.01mol)加入50ml四氢呋喃与乙醇(v
四氢呋喃
:v
乙醇
=3:1)的混合液中搅拌溶解,溶清后将体系升温至40~45℃加入naoh(0.32g,0.008mol),加毕,继续保温搅拌反应1h,反应完毕后降至室温,加入50ml去离子水和50ml乙酸乙酯,震荡摇匀,静置分液,水相用25ml乙酸乙酯提取一遍,合并有机相,分别用饱和碳酸氢钠溶液和饱和食盐水洗涤分液,有机相用无水硫酸钠干燥后减压浓缩,柱层析分离(洗脱剂先用v
石油醚
:v
乙酸乙酯
=1:1洗脱前杂,后用乙酸乙酯洗脱)得化合物i,收率88.5%,hplc纯度99.97%。
[0030]
实施例2
[0031]
室温下,将依维莫司(10.00g,0.01mol)加入50ml三氯甲烷与乙醇(v
三氯甲烷
:v
乙醇
=4:1)的混合液中搅拌溶解,溶清后将体系升温至40~45℃加入na2co3(0.85g,0.008mol),加毕,继续保温搅拌反应1h,反应完毕后降至室温,加入50ml去离子水和50ml二氯甲烷,震荡摇匀,静置分液,水相用25ml二氯甲烷提取一遍,合并有机相,分别用饱和碳酸氢钠溶液和饱和食盐水洗涤分液,有机相用无水硫酸钠干燥后减压浓缩,柱层析分离(洗脱剂先用v
石油醚
:v
乙酸乙酯
=1:1洗脱前杂,后用乙酸乙酯洗脱)得化合物i,收率86.5%,hplc纯度99.84%。
[0032]
实施例3
[0033]
室温下,将依维莫司(10.00g,0.01mol)加入50ml四氢呋喃与乙醇(v
四氢呋喃
:v
乙醇
=2:1)的混合液中搅拌溶解,溶清后将体系升温至30~35℃,加入naoh(0.2g,0.005mol),加毕,继续保温搅拌反应1h,反应完毕后降至室温,加入50ml去离子水和50ml乙酸乙酯,震荡摇匀,静置分液,水相用25ml乙酸乙酯提取一遍,合并有机相,分别用饱和碳酸氢钠溶液和饱和食盐水洗涤分液,有机相用无水硫酸钠干燥后减压浓缩,柱层析分离(洗脱剂先用v
石油醚
:v
乙酸乙酯
=1:1洗脱前杂,后用乙酸乙酯洗脱)得化合物i,收率85.2%,hplc纯度99.60%。
[0034]
实施例4
[0035]
室温下,将化合物42-o-(2-羟基)乙基雷帕霉素(10.00g,0.01mol)加入50ml1,4-二氧六环与乙醇(v
1,4-二氧六环
:v
乙醇
=2:1)的混合液中搅拌溶解,于60~65℃加入naoh(0.60g,0.015mol),加毕,继续室温搅拌反应1h,反应完毕后,加入50ml去离子水和50ml乙酸乙酯,震荡摇匀,静置分液,水相用25ml乙酸乙酯提取一遍,合并有机相,分别用饱和碳酸氢钠溶液和饱和食盐水洗涤分液,有机相用无水硫酸钠干燥后减压浓缩,柱层析分离(洗脱剂先用v
石油醚
:v
乙酸乙酯
=1:1洗脱前杂,后用乙酸乙酯洗脱)得化合物i,收率84.7%,hplc纯度99.54%。
[0036]
实施例5
[0037]
室温下,将化合物42-o-(2-羟基)乙基雷帕霉素(10.00g,0.01mol)加入50ml无水乙醇中搅拌溶解,溶清后将体系升温至25~30℃,加入naoh(0.012g,0.003mmol),加毕,继续保温搅拌反应1h,反应完毕后降至室温,加入50ml去离子水和50ml乙酸乙酯,震荡摇匀,静置分液,水相用25ml乙酸乙酯提取一遍,合并有机相,分别用饱和碳酸氢钠溶液和饱和食盐水洗涤分液,有机相用无水硫酸钠干燥后减压浓缩,柱层析分离(洗脱剂先用v
石油醚
:v
乙酸乙酯
=1:1洗脱前杂,后用乙酸乙酯洗脱)得化合物i,收率80.2%,hplc纯度98.93%。
[0038]
实施例6
[0039]
室温下,将化合物42-o-(2-羟基)乙基雷帕霉素(10.00g,0.01mol)加入50ml1.4-二氧六环与乙醇(v
1.4-二氧六环
:v
乙醇
=1:1)的混合液中搅拌溶解,升温至65~70℃,加入k2co3(2.35g,0.017mol),加毕,继续室温搅拌反应1h,反应完毕后,加入50ml去离子水和50ml乙酸乙酯,震荡摇匀,静置分液,水相用25ml乙酸乙酯提取一遍,合并有机相,分别用饱和碳酸氢钠溶液和饱和食盐水洗涤分液,有机相用无水硫酸钠干燥后减压浓缩,柱层析分离(洗脱剂先用v
石油醚
:v
乙酸乙酯
=1:1洗脱前杂,后乙酸乙酯洗脱)得化合物i,收率79.6%,hplc纯度98.86%。
[0040]
对比实施例
[0041]
将依维莫司(0.2g)溶于无水乙醇(20ml),加入0.1n的盐酸(10ml),20℃~30℃下搅拌24小时,加入200ml水,乙酸乙酯萃取(150ml
×
3),合并有机相,饱和碳酸氢钠洗1次,饱和食盐水洗涤2次。无水na2so4干燥,过滤,滤液25℃真空浓缩至干。粗品经制备液相纯化得目标产物乙基化杂质,收率42%,hplc纯度93.2%,异构体杂质4.25%。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1