生产CAR-T细胞的方法与流程
生产car-t细胞的方法
1.相关申请的交叉引用
2.本技术要求于2019年11月13日提交的美国临时专利申请号62/934,999的优先权权益,该临时专利申请特此通过援引以其全文并入。
背景技术:
3.嵌合抗原受体(car)t-细胞疗法已经在治疗血液癌中显示出有前景的治疗效果。通常,car-t细胞通过患者免疫细胞(自体)或来自不相关人类供体的免疫细胞(同种异体)的基因工程而产生。高质量、临床级car-t细胞的制备是该技术广泛应用的先决条件。因此,非常感兴趣的是开发用于大规模制备car-t细胞的有效生产方法。
技术实现要素:
4.本披露至少部分地基于用于生产表达嵌合抗原受体(car)的基因工程化t细胞的方法的开发,这些方法提供优于常规生产方法的几种改进。此类改进包括但不限于本文所述的基因修饰的一致性和效率的改进(例如,三重基因组编辑的一致性和效率的改进),这允许产生临床上有用的car t细胞疗法的稳健供应。
5.因此,本披露的一个方面提供了一种用于生产基因工程化t细胞的方法,该方法包括(i)提供第一t细胞群体;(ii)向该第一t细胞群体中引入包含第一cas9酶和靶向cd70基因的第一引导rna(grna)的第一核糖核蛋白(rnp)复合物以产生第二t细胞群体,其中该第二t细胞群体包含具有破坏的该cd70基因的t细胞;(iii)向该第二t细胞群体中引入包含第二cas9酶和靶向t细胞受体α链恒定区(trac)基因的第二grna的第二rnp复合物以及包含第三cas9酶和靶向β-2微球蛋白(β2m)基因的第三grna的第三rnp复合物以产生第三t细胞群体,其中该第三t细胞群体包含具有破坏的该cd70基因、破坏的该trac基因和破坏的该β2m基因的激活的t细胞;(iv)将该第三t细胞群体与腺相关病毒(aav)载体一起温育以产生第四t细胞群体,其中该aav载体包含编码嵌合抗原受体(car)的核酸序列并且其中该核酸序列的两侧有该trac基因的同源序列,并且其中该第四t细胞群体包含表达该car并具有破坏的该cd70基因、破坏的该trac基因和破坏的该β2m基因的激活的t细胞;(v)扩增该第四t细胞群体,从而产生扩增的t细胞群体;(vi)从该扩增的t细胞群体中去除tcrαβ
+
t细胞以产生基因工程化t细胞群体,其中该基因工程化t细胞群体包含表达该car并具有破坏的该cd70基因、破坏的该trac基因和破坏的该β2m基因的激活的t细胞;以及(vii)收获该基因工程化t细胞群体。
6.在一些实施例中,第一t细胞群体来源于从人血细胞富集的冻存的t细胞。在一些实施例中,第一t细胞群体通过包括以下步骤的过程制备:(a)从人类供体获得血细胞;以及(b)从这些血细胞富集cd4
+
t细胞和/或cd8
+
t细胞。在一些实施例中,使用与抗cd4和/或抗cd8抗体缀合的磁珠进行步骤(b)。在一些实施例中,第一t细胞群体具有至少约80%的细胞活力和/或至少约80%的cd4
+
和cd8
+
t细胞的纯度。在一些实施例中,方法还包括(c)冻存步骤(b)中产生的富集的cd4
+
t细胞和cd8
+
t细胞。
7.在一些实施例中,通过电穿孔进行步骤(ii)。在一些实施例中,第一cas9酶的浓度为约0.15mg/ml并且靶向cd70基因的第一grna的浓度为约0.16mg/ml。在一些实施例中,步骤(ii)中的细胞浓度为约100x106个细胞/ml至约400x106个细胞/ml。在一些实施例中,步骤(ii)中的细胞浓度为约100x106个细胞/ml至约350x106个细胞/ml。在一些实施例中,步骤(ii)中的细胞浓度为约300x106个细胞/ml。
8.在一些实施例中,这些方法还包括在步骤(ii)之后且在步骤(iii)之前,在细胞培养容器中在存在t细胞激活剂的情况下温育该第二t细胞群体以产生激活的t细胞群体的步骤,其中该激活的t细胞群体包含具有破坏的该cd70基因的激活的t细胞。该t细胞激活剂可包括cd3激动剂和cd28激动剂,并且其中该cd3激动剂和cd28激动剂附着于纳米基质颗粒。在细胞培养容器中在存在t细胞激活剂的情况下温育第二t细胞群体可以在约2x106/cm2的细胞接种密度和约2x106个细胞/ml的细胞浓度下进行约72小时。在一些实施例中,混合物中t细胞激活剂与培养基的比率为约1:12.5(v/v)。在其他实施例中,本文披露的方法还可包括在存在t细胞激活剂的情况下温育第二t细胞群体后,稀释激活的t细胞群体中的t细胞激活剂以减少激活并允许细胞在步骤(iii)之前恢复。
9.在一些实施例中,通过电穿孔进行步骤(iii)。在一些实施例中,步骤(iii)涉及一个电穿孔事件。在一些实施例中,在该一个电穿孔事件中将第二rnp复合物和第三rnp复合物引入到激活的t细胞中。在一些实施例中,第二rnp复合物中的第二cas9酶的量与第三rna复合物中的第三cas9酶的量相同。在一些实施例中,第二cas9酶的浓度为约0.3mg/ml,第三cas9酶的浓度为约0.3mg/ml,靶向trac基因的第二grna的浓度为约0.08mg/ml,并且靶向β2m基因的第三grna的浓度为约0.2mg/ml。在一些实施例中,步骤(iii)中的细胞浓度为约100x106个细胞/ml至约400x106个细胞/ml。在一些实施例中,步骤(iii)中的细胞浓度为约300x106个细胞/ml。在其他实施例中,步骤(iii)(例如,电穿孔)中使用的每个容器中的总细胞数可为约5x108至约2.5x109个细胞,例如约7x108个细胞。在一些实例中,可在步骤(iii)(例如,电穿孔)中使用多个容器,例如约5至10个容器。在具体实例中,可在步骤(iii)中使用多达7个容器,这些容器可含有例如用于电穿孔的约1.5x109至约3x109个细胞(例如,约2.1x109个细胞或约2.7x109个细胞)。
10.在一些实施例中,aav载体具有约10,000至约80,000的感染复数(moi)值。在一些实施例中,aav载体的moi为约20,000。在一些实施例中,aav载体是aav血清型6(aav6)载体。
11.在一些实施例中,通过在细胞培养容器中以约2x105个细胞/cm2至约5x105个细胞/cm2的接种密度培养第四t细胞群体约7天至约12天来进行步骤(v)。在一些实施例中,可将第四t细胞群体以约150,000个细胞/cm2至约600,000个细胞/cm2的接种密度接种于细胞培养容器中。在一些实施例中,以约3x105个细胞/cm2至约5x105个细胞/cm2的接种密度培养第四t细胞群体。在一些实施例中,细胞培养容器是在不更换培养基的情况下允许细胞扩增约10天至约12天的静态细胞培养容器(在本文中也可互换地称为静态培养容器)。
12.在一些实施例中,通过使扩增的细胞与其上固定有抗tcrαβ抗体的珠接触并收集未结合的细胞来进行步骤(vi)。
13.在一些实施例中,第一cas9酶、第二cas9酶和/或第三cas9酶是来自酿脓链球菌(streptococcus pyogenes)的cas9(spcas9)的cas9酶。在一些实施例中,第一cas9酶、第二cas9酶和第三cas9酶是相同的。在一些实施例中,第一cas9酶、第二cas9酶和第三cas9酶包
含seq id no:1的氨基酸序列。
14.在一些实施例中,靶向cd70基因的第一grna包含seq id no:4的间隔子序列。在一些实施例中,靶向cd70基因的第一grna包含seq id no:2的核苷酸序列。
15.在一些实施例中,靶向trac基因的第二grna包含seq id no:8的间隔子序列。在一些实施例中,靶向trac基因的第二grna包含seq id no:6的核苷酸序列。
16.在一些实施例中,靶向β2m基因的第三grna包含seq id no:12的间隔子序列。在一些实施例中,靶向β2m基因的第三grna包含seq id no:10的核苷酸序列。
17.在一些实施例中,第一grna、第二grna、第三grna和/或它们的组合包含一个或多个2
′‑
o-甲基硫代磷酸酯修饰。
18.在一些实施例中,car包含靶向癌抗原的胞外结构域、跨膜结构域、共刺激结构域和cd3ζ细胞质信号传导结构域。在一些实施例中,胞外结构域包含单链可变片段(scfv),跨膜结构域来源于cd8a,并且/或者共刺激结构域来源于4-1bb。在一些实施例中,scfv片段结合cd70。在一些实施例中,car包含seq id no:46的氨基酸序列。
19.因此,本披露的一个方面提供了一种用于生产基因工程化t细胞的方法,该方法包括(i)提供第一t细胞群体;(ii)向该第一t细胞群体中引入包含第一cas9酶和靶向cd70基因的第一引导rna(grna)的第一核糖核蛋白(rnp)复合物以产生第二t细胞群体,其中该第二t细胞群体包含具有破坏的该cd70基因的t细胞;(iii)在细胞培养容器中在存在t细胞激活剂的情况下温育该第二t细胞群体以产生第三t细胞群体,其中该第三t细胞群体包含具有破坏的该cd70基因的激活的t细胞;(iv)向该第三t细胞群体中引入包含第二cas9酶和靶向t细胞受体α链恒定区(trac)基因的第二grna的第二rnp复合物以及包含第三cas9酶和靶向β-2微球蛋白(β2m)基因的第三grna的第三rnp复合物以产生第四t细胞群体,其中该第四t细胞群体包含具有破坏的该cd70基因、破坏的该trac基因和破坏的该β2m基因的激活的t细胞;(v)将该第四t细胞群体与腺相关病毒(aav)载体一起温育以产生第五t细胞群体,其中该aav载体包含编码嵌合抗原受体(car)的核酸序列并且其中该核酸序列的两侧有该trac基因的同源序列,并且其中该第五t细胞群体包含表达该car并具有破坏的该cd70基因、破坏的该trac基因和破坏的该β2m基因的激活的t细胞;(vi)扩增该第五t细胞群体,从而产生扩增的t细胞群体;(vii)从该扩增的t细胞群体中去除tcrαβ
+
t细胞以产生基因工程化t细胞群体,其中该基因工程化t细胞群体包含表达该car并具有破坏的该cd70基因、破坏的该trac基因和破坏的该β2m基因的激活的t细胞;以及(viii)收获该基因工程化t细胞群体。
20.在一些实施例中,一种基因工程化t细胞群体,其通过本文所述的方法产生。
21.本发明的一个或多个实施例的细节在以下说明书中阐述。根据以下附图和对几个实施例的详细描述,并且还根据所附权利要求书,本发明的其他特征或优点将变得显而易见。
附图说明
22.图1是示出在小规模生产过程中制备的t细胞的编辑后t细胞扩增的图表。在括号中指示了rnp复合物。2d:t细胞激活2天(48小时);3d:t细胞激活3天(72小时);1x ep:单次电穿孔;2x ep:两步电穿孔。
23.图2a至图2b包括示出单次电穿孔或两步电穿孔对易位率的影响的图表。图2a:示出11种所指示的易位的易位百分比的图表。图2b:示出8种所指示的易位的易位百分比的图表。
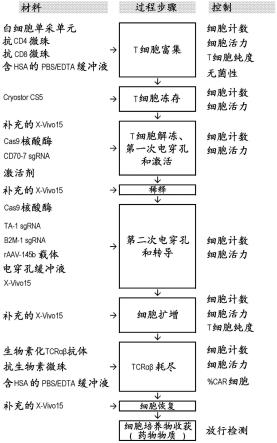
24.图3a至图3b包括用于制备ctx130 t细胞的方法的流程图,这些ctx130 t细胞表达抗cd70 car并具有基因破坏的cd70、β2m和trac基因。图3a包括根据本文所述技术的一些实施例的用于制备表达抗cd70car的t细胞的说明性生产过程的流程图。car:嵌合抗原受体;edta:乙二胺四乙酸;hsa:人血清白蛋白;il:白介素;pbs:磷酸盐缓冲盐水;raav:重组腺相关病毒;sgrna:单引导核糖核酸;tcrαβ:t细胞受体α链和t细胞受体β链;补充的x-vivo
tm 15:含5%男性人类血清ab、100iu/ml rhil-2和100iu/ml rhil-7的x-vivo
tm 15。图3b包括根据本文所述技术的一些实施例的用于制备药物产品的说明性生产过程的流程图,该药物产品包含表达抗cd70 car的t细胞。
具体实施方式
25.本披露至少部分地基于用于产生car-t细胞,特别是同种异体car-t细胞的改进的生产过程的开发,包括用于生产过程的一个或多个步骤的改进的条件。本文披露的改进的生产过程引起至少以下有利结果:
26.(a)因本文提供的改进的t细胞激活条件而得到电穿孔后增加的%car
+
表达和减少的细胞损失。
27.(b)因本文提供的激活的t细胞条件的改进的crispr-cas9介导的基因编辑而得到t细胞中β2m基因破坏的改进的一致性和改进的效率。
28.(c)因本文提供的改进的t细胞电穿孔条件而得到较低易位率。
29.(d)因本文所述改进的生产过程提供的减少的制备时间和减少的制备成本而得到car t细胞疗法的增加的供应。
30.(e)因使用本文所述改进的生产过程制备均匀且高质量的car t疗法而得到所生产的药物产品的减少的可变性。
31.(f)简化aav转导条件,同时在t细胞中保持高car表达水平。
32.因此,本文提供了用于生产表达car构建体(诸如靶向癌抗原例如cd70的car构建体)并且具有被敲除的cd70、trac和β2m基因的基因工程化t细胞的方法。通过本文所述的方法产生的基因工程化t细胞群体及其治疗用途也在本披露的范围内。
33.i.生产基因工程化t细胞
34.本披露的各方面提供了用于生产基因工程化t细胞的方法,这些基因工程化t细胞包含破坏的分化簇70(cd70)基因、破坏的β2微球蛋白(β2m)基因和破坏的t细胞受体α链恒定区(trac)基因以及编码嵌合抗原受体(car)的插入的核酸。
35.cd70基因的破坏防止了基因工程化t细胞生产期间的细胞间相互杀伤。替代性地或作为补充,cd70基因的破坏实现了基因工程化t细胞的健康和功能提升(例如,增殖延长、耗竭减少)。β2m基因和trac基因的破坏使基因工程化t细胞无同种异体反应性并且适合同种异体移植。编码car的核酸的插入使得基因工程化t细胞能够在其表面上表达car,该car在此将基因工程化t细胞靶向癌细胞。
36.因此,在一些实施例中,本文披露的用于生产基因工程化t细胞的方法涉及使用
crispr-cas9基因编辑来破坏cd70、trac和β2m基因的表达,并且使用腺相关病毒(aav)转导来插入编码car的核酸。
37.总体而言,本文披露的用于生产car-t细胞的方法可包括:(i)从合适的人免疫细胞来源富集cd4
+
/cd8
+
t细胞,(ii)激活富集的cd4
+
/cd8
+
t细胞;(iii)对激活的t细胞进行基因工程化以产生具有破坏的cd70、trac和β2m基因的car-t细胞;以及收获基因工程化t细胞以用于治疗用途。当需要时,可通过冻存来储存富集的cd4
+
/cd8
+
t细胞以备将来使用。替代性地或作为补充,可在收获前在体外扩增基因工程化t细胞。可从由此产生的car-t细胞群体中耗尽tcrαβ
+
t细胞。
38.(i)t细胞富集
39.本文披露的任何生产方法可使用人血细胞作为原材料。例如,可以使用本领域技术人员已知的技术诸如沉降(例如,ficoll
tm
分离)从收集自受试者的单位血液中获得t细胞。替代性地,用于制备基因工程化t细胞的t细胞可通过体外分化来源于干细胞(例如hsc或ipsc)。在一些实施例中,可从单独人类供体获得血细胞。在其他实施例中,可从多个人类供体(例如,2、3、4或5个人类供体)获得血细胞。
40.在一些实例中,可使用来自合适人类供体的白细胞单采样品。如本领域已知的,白细胞单采样品是从外周血收集的富集的白细胞单采产物。它通常含有多种血细胞,包括单核细胞、淋巴细胞、血小板、血浆和红细胞。人类供体优选是健康的人类供体。例如,可对人类供体候选者进行hbv、hcv、hiv、htlv、wnv、克氏锥虫和/或cmv筛查。在筛查中显示阴性结果的人类受试者可用作血细胞的供体。
41.可用于本发明方法的t细胞的来源没有特别限制。在一些实施例中,来自t细胞库的t细胞可用作本文披露的任何生产方法中的原材料。t细胞库可包含具有某些基因(例如,参与细胞自我更新、细胞凋亡和/或t细胞耗竭或复制性衰老的基因)的基因编辑的t细胞,以改善细胞培养中的t细胞持久性。t细胞库可由真正的t细胞产生,例如,非转化t细胞、终末分化t细胞、具有稳定基因组的t细胞和/或依赖于细胞因子和生长因子进行增殖和扩增的t细胞。替代性地,t细胞库可由前体细胞产生,诸如造血干细胞(例如,ipsc)(例如,体外培养)。在一些实例中,t细胞库中的t细胞可包含参与细胞自我更新的一个或多个基因、参与细胞凋亡的一个或多个基因和/或参与t细胞耗竭的一个或多个基因的基因编辑,以便破坏或减少此类基因的表达,从而提高培养中的持久性。t细胞库中编辑的基因的实例包括但不限于tet2、fas、cd70、regnase-1或它们的组合。与未编辑的t对应物相比,t细胞库中的t细胞可具有培养中增强的扩增能力、增强的增殖能力、更大的t细胞激活和/或降低的细胞凋亡水平。
42.可以使用常规方法或本文披露的方法从人血细胞中富集合适的t细胞。用于制备基因工程化t细胞的t细胞可表达一种或多种t细胞标志物,包括但不限于cd4
+
、cd8
+
或它们的组合。在一些实施例中,可以从人血细胞中富集cd4
+
t细胞。在其他实施例中,可以富集cd8
+
t细胞。在具体实例中,cd4
+
和cd8
+
t细胞均纯化自人血细胞。
43.可以使用本领域已知的任何方法或本文披露的方法,例如使用能够结合靶t细胞的特异性细胞表面生物标志物的抗体(例如cd4特异性抗体和/或cd8特异性抗体),从合适的血细胞来源(例如本文描述的那些)分离cd4
+
t细胞和/或cd8
+
t细胞。在一些实施例中,富集cd4
+
t细胞和cd8
+
t细胞可以使用与磁珠缀合的抗cd4和抗cd8抗体进行。可以将包含cd4
+
和cd8
+
t细胞的细胞群体与此类磁珠一起在合适的条件下温育一段合适的时间,从而允许靶t细胞通过与磁珠缀合的抗体来结合磁珠。可以洗涤未结合的细胞,并且可以使用常规方法收集与珠结合的cd4
+
和cd8
+
t细胞。
44.可以在常规实践后评价富集的t细胞(例如,cd4
+
t细胞和cd8
+
t细胞)的特征,例如细胞活力和/或靶t细胞的纯度。在一些实施例中,来自本文披露的富集步骤的t细胞群体可具有至少约80%(例如,至少约85%、至少约90%、至少约95%或更高)的细胞活力。替代性地或作为补充,富集的t细胞群体可具有至少约80%,例如至少约85%、至少约90%、至少约95%、至少约97%、约98%或更高的靶t细胞(例如cd4
+
和/或cd8
+
t细胞)的纯度。替代性地或作为补充,富集的t细胞群体可具有至少约70%,例如至少约75%、至少约80%、至少约85%、至少约90%、至少约95%、至少约97%、约98%或更高的靶t细胞(例如cd4
+
和/或cd8
+
t细胞)的纯度。
45.术语“约”或“大约”意指在本领域普通技术人员确定的特定值的可接受误差范围内,这将部分地取决于如何测量或确定该值,即测量系统的限制。例如,根据本领域的实践,“约”可以意指在可接受的标准偏差内。替代性地,“约”可以意指给定值的至多
±
20%,优选至多
±
10%,更优选至多
±
5%,且更优选至多
±
1%的范围。替代性地,特别是对于生物系统或过程而言,该术语可以意指在数值的一个数量级内,优选在2倍内。在本技术和权利要求书中描述了特定值的情况下,除非另有说明,否则术语“约”是隐含的并且在该上下文中意指在特定值的可接受误差范围内。
46.富集的t细胞群体(其也在本披露的范围内)可以如本文披露的那样立即用于进一步处理。替代性地,富集的t细胞群体可以在合适的条件下储存以备将来使用,例如通过冻存来储存。在进一步处理之前,冻存的t细胞可以按照常规程序解冻。可以评估解冻的细胞的细胞活力以确定解冻的细胞是否适合进一步处理。
47.(ii)富集的t细胞的crispr-cas9介导的基因编辑
48.可以通过例如crispr-cas9基因编辑技术对通过本文披露的任何程序制备的富集的t细胞进行基因编辑以敲除cd70。在第一电穿孔步骤中敲除cd70基因,随后在第二电穿孔步骤中敲除trac和β2m基因显著增加了编辑效率并减少了基因编辑期间产生的易位数目。参见以下实例。
49.cd70基因编码肿瘤坏死因子超家族的成员,并且其表达限于激活的t淋巴细胞和b淋巴细胞以及成熟的树突状细胞。cd70通过与其配体cd27相互作用而参与肿瘤细胞和调节性t细胞的存活。cd70基因的破坏使基因工程化t细胞生产期间的细胞间相互杀伤的风险最小化,并且实现了生产的基因工程化t细胞的健康和功能提升。
50.crispr-cas9介导的基因编辑系统
51.crispr-cas9系统是原核生物中天然存在的防御机制,其已被重新用作用于基因编辑的rna引导的dna靶向平台。它依赖于dna核酸酶cas9和两个非编码rna(crisprrna(crrna)和反式激活rna(tracrrna))来靶向dna的切割。crispr是规律间隔成簇短回文重复序列的首字母缩略词,是在细菌和古细菌基因组中发现的dna序列家族,其包含与先前暴露于细胞的外源dna(例如,由感染或攻击原核生物的病毒暴露于细胞的外源dna)相似的dna片段(间隔子dna)。这些dna片段被原核生物用来在重新引入后,例如在随后的攻击中从相似的病毒中检测和破坏相似的外源dna。crispr基因座的转录导致包含间隔子序列的rna分
子的形成,该rna分子缔合并靶向cas(crispr相关)蛋白以能够识别和剪切外源(外源dna)。已经描述了许多类型和种类的crispr/cas系统(参见例如,koonin等人,(2017)curr opin microbiol[微生物学当前观点]37:67-78)。
[0052]
crrna通过典型地与靶dna中的20个核苷酸(nt)序列的沃森-克里克碱基配对来驱动crispr-cas9复合物的序列识别和特异性。改变crrna中5’20nt的序列可将crispr-cas9复合物靶向特定基因座。如果靶序列后面是特定的短dna序列(序列为ngg)作为原型间隔子相邻基序(pam),crispr-cas9复合物仅结合包含与crrna的前20nt序列匹配的dna序列。
[0053]
tracrrna与crrna的3’末端杂交形成rna双链体结构,该双链体结构与cas9内切核酸酶结合形成催化活性的crispr-cas9复合物,其然后可以切割靶dna。
[0054]
一旦crispr-cas9复合物在靶位点与dna结合,cas9酶内的两个独立的核酸酶结构域各自切割pam位点上游的dna链之一,从而留下双链断裂(dsb),在这里dna的两条链以碱基对(平末端)终止。
[0055]
crispr-cas9复合物在特定靶位点处与dna结合并形成位点特异性dsb之后,下一个关键步骤是修复dsb。细胞使用两种主要的dna修复途径来修复dsb:非同源末端连接(nhej)和同源定向修复(hdr)。
[0056]
nhej是一种稳健的修复机制,在包括非分裂细胞在内的大多数细胞类型中显现出高活性。nhej容易出错,并且通常会在dsb的位点导致在一个到几百个核苷酸之间的去除或添加,尽管此类修饰典型地《20nt。产生的插入和缺失(插缺)可以破坏基因的编码或非编码区域。替代性地,hdr使用内源性或外源性提供的长段同源供体dna来以高保真度修复dsb。hdr仅在分裂的细胞中有效,并且在大多数细胞类型中以相对较低的频率发生。在本披露的许多实施例中,nhej被作为自发的修复来利用。
[0057]
(a)cas9
[0058]
在一些实施例中,cas9(crispr相关蛋白9)核酸内切酶用于crispr方法中,该方法用于制备本文披露的基因工程化t细胞。cas9酶可以是酿脓链球菌中的一种,尽管也可以使用其他cas9同源物。应当理解,如本文所提供的,可以使用野生型cas9或可以使用cas9的修饰版本(例如,cas9的进化版本,或cas9直向同源物或变体)。在一些实施例中,cas9包含酿脓链球菌衍生的cas9核酸酶蛋白,其已被工程化为包括c末端和n末端sv40大t抗原核定位序列(nls)。所得的cas9核酸酶(snls-spcas9-snls)是162kda蛋白,其通过重组大肠杆菌发酵产生并通过色谱法纯化。spcas9氨基酸序列可作为uniprot登录号q99zw2找到,其在本文中作为seq id no:1提供。
[0059]
(b)引导rna(grna)
[0060]
如本文所述,crispr-cas9介导的基因编辑包括引导rna或grna的使用。如本文所用,“grna”是指基因组靶向核酸,其可以将cas9定向至cd70基因或trac基因或β2m基因内的特定靶序列,以在特定靶序列处进行基因编辑。引导rna至少包含与靶基因内用于进行编辑的靶核酸序列杂交的间隔子序列,以及crispr重复序列。
[0061]
seq id no:2中提供了靶向cd70基因的示例性grna。还参见2019年5月10日提交的国际申请号pct/ib 2019/000500(现已公开为wo2019/215500),该国际申请的相关披露内容通过援引并入本文用于本文所引用的主题和目的。可以使用位于第19号染色体上的cd70基因序列(grch38:第19号染色体:6,583,183-6,604,103;ensembl;ensg00000125726)设计
其他grna序列。
[0062]
在一些实施例中,靶向cd70基因组区域的grna和cas9在cd70基因组区域中产生断裂,导致cd70基因中的插入缺失,其破坏mrna或蛋白质的表达。在一些实施例中,靶向cd70基因组区域的grna在cd70基因中产生插入缺失,该cd70基因包含选自表11中的序列的至少一个核苷酸序列。在一些实施例中,靶向cd70基因组区域的grna(seq id no:2)在cd70基因中产生插入缺失,该cd70基因包含选自表11中的序列的至少一个核苷酸序列。
[0063]
seq id no:6中提供了靶向trac基因的示例性grna。还参见2018年5月11日提交的国际申请号pct/ib 2018/001619(已公开为wo2019/097305a2),该国际申请的相关披露内容通过援引并入本文用于本文所引用的主题和目的。可以使用位于第14号染色体上的trac基因序列(grch38:第14号染色体:22,547,506-22,552,154;ensembl;ensg00000277734)设计其他grna序列。
[0064]
在一些实施例中,靶向trac基因组区域的grna和cas9在trac基因组区域中产生断裂,导致trac基因中的插入缺失,其破坏mrna或蛋白质的表达。在一些实施例中,靶向trac基因组区域的grna在trac基因中产生插入缺失,该trac基因包含选自表9中的序列的至少一个核苷酸序列。在一些实施例中,靶向trac基因组区域的grna(seq id no:6)在trac基因中产生插入缺失,该trac基因包含选自表9中的序列的至少一个核苷酸序列。
[0065]
seq id no:10中提供了靶向β2m基因的示例性grna。还参见2018年5月11日提交的国际申请号pct/ib 2018/001619(已公开为wo2019/097305a2),该国际申请的相关披露内容通过援引并入本文用于本文所引用的目的和主题。可以使用位于第15号染色体上的β2m基因序列(grch38坐标:第15号染色体:44,711,477-44,718,877;ensembl:ensg00000166710)设计其他grna序列。
[0066]
在一些实施例中,靶向β2m基因组区域的grna和rna引导的核酸酶在β2m基因组区域中产生断裂,导致β2m基因中的插入缺失,其破坏mrna或蛋白质的表达。在一些实施例中,靶向β2m基因组区域的grna在β2m基因中产生插入缺失,该β2m基因包含选自表10中的序列的至少一个核苷酸序列。在一些实施例中,靶向β2m基因组区域的grna(seq id no:10)在β2m基因中产生插入缺失,该β2m基因包含选自表10中的序列的至少一个核苷酸序列。
[0067]
在ii型系统中,grna还包含称为tracrrna序列的第二rna。在ii型grna中,crispr重复序列和tracrrna序列彼此杂交形成双链体。在v型grna中,crrna形成双链体。在这两个系统中,双链体都结合定点多肽,使得引导rna和定点多肽形成复合物。在一些实施例中,靶向基因组的核酸由于其与定点多肽的缔合而为复合物提供了靶特异性。因此,靶向基因组的核酸定向定点多肽的活性。
[0068]
如本领域普通技术人员所理解的,每个定向rna设计为包括与其基因组靶序列互补的间隔子序列。参见jinek等人,science[科学],337,816-821(2012)和deltcheva等人,nature[自然],471,602-607(2011)。
[0069]
在一些实施例中,靶向基因组的核酸(例如,grna)是双分子引导rna。在一些实施例中,靶向基因组的核酸(例如,grna)是单分子引导rna。
[0070]
双分子引导rna包含两条链的rna分子。第一条链在5'至3'方向上包含任选的间隔子延伸序列、间隔子序列和最小crispr重复序列。第二条链包含最小tracrrna序列(与最小crispr重复序列互补)、3’tracrrna序列和任选的tracrrna延伸序列。
[0071]
ii型系统中的单分子引导rna(称为“sgrna”)在5'至3'方向上包含任选的间隔子延伸序列、间隔子序列、最小crispr重复序列、单分子引导接头、最小tracrrna序列、3’tracrrna序列和任选的tracrrna延伸序列。任选的tracrrna延伸序列可以包含为引导rna贡献另外的功能(例如,稳定性)的元件。单分子引导接头将最小crispr重复序列和最小tracrrna序列连接起来以形成发夹结构。任选的tracrrna延伸包括一个或多个发夹。v型系统中的单分子引导rna在5'至3'方向上包含最小crispr重复序列和间隔子序列。
[0072]“靶序列”在与pam序列相邻的靶基因中,并且是要被cas9修饰的序列。“靶序列”在“靶核酸”中的所谓的pam链上,该靶核酸是包含pam链和互补的非pam链的双链分子。本领域技术人员认识到,grna间隔子序列与位于目的靶核酸的非pam链中的互补序列杂交。因此,grna间隔子序列是靶序列的rna等同物。
[0073]
例如,如果cd70靶序列是5
′‑
gctttggtcccattggtcgc-3
′
(seq id no:15),则grna间隔子序列是5
′‑
gcuuuggucccauuggucgc-3
′
(seq id no:5)。在另一个实例中,如果trac靶序列是5
′‑
agagcaacagtgctgtggcc-3
′
(seq id no:17),则grna间隔子序列是5
′‑
agagcaacagugcuguggcc-3
′
(seq id no:9)。在又一个实例中,如果β2m靶序列是5
′‑
gctactctctctttctggcc-3
′
(seq id no:19),则grna间隔子序列是5
′‑
gcuacucucucuuucuggcc-3
′
(seq id no:13)。grna的间隔子经由杂交(即,碱基配对)以序列特异性方式与目的靶核酸相互作用。因此,间隔子的核苷酸序列根据目的靶核酸的靶序列而变化。
[0074]
在本文的crispr/cas系统中,将间隔子序列设计成与靶核酸的区域杂交,该区域位于该系统中使用的cas9酶可识别的pam的5'。间隔子可以与靶序列完全匹配或可以有错配。每个cas9酶都有特定的pam序列,使得该酶识别靶dna。例如,酿脓链球菌识别靶核酸中的包含序列5'-nrg-3'的pam,其中r包含a或g,其中n是任何核苷酸并且n紧邻由间隔子序列靶向的靶核酸序列的3'。
[0075]
在一些实施例中,靶核酸序列的长度是20个核苷酸。在一些实施例中,靶核酸的长度小于20个核苷酸。在一些实施例中,靶核酸的长度超过20个核苷酸。在一些实施例中,靶核酸具有至少:5、10、15、16、17、18、19、20、21、22、23、24、25、30或更多个核苷酸长度。在一些实施例中,靶核酸至多具有:5、10、15、16、17、18、19、20、21、22、23、24、25、30或更多个核苷酸长度。在一些实施例中,靶核酸序列具有20个紧邻pam第一个核苷酸的5'的碱基。例如,在包含5'-nnnnnnnnnnnnnnnnnnnnnrg-3'的序列中,靶核酸可以是对应于该多个n的序列,其中n可以是任何核苷酸,并且该加下划线的nrg序列是酿脓链球菌pam。
[0076]
grna中的间隔子序列是定义目标靶基因的靶序列(例如,dna靶序列,诸如基因组靶序列)的序列(例如,20个核苷酸的序列)。seq id no:4中提供了靶向cd70基因的grna的示例性间隔子序列。seq id no:8中提供了靶向trac基因的grna的示例性间隔子序列。seq id no:12中提供了靶向β2m基因的grna的示例性间隔子序列。
[0077]
本文披露的引导rna可以经由crrna中的间隔子序列靶向任何目的序列。在一些实施例中,引导rna的间隔子序列与靶基因中的靶序列之间的互补程度可以是约60%、65%、70%、75%、80%、85%、90%、95%、97%、98%、99%或100%。在一些实施例中,引导rna的间隔子序列与靶基因中的靶序列是100%互补的。在其他实施例中,引导rna的间隔子序列和靶基因中的靶序列可以包含多达10个错配,例如,多达9个、多达8个、多达7个、多达6个、
多达5个、多达4个、多达3个、多达2个或多达1个错配。
[0078]
可以如本文所提供的那样使用的grna的非限制性实例提供于2018年5月11日提交的pct/ib 2018/001619(已公开为wo 2019/097305 a2)和2019年5月10日提交的pct/ib 2019/000500(现已公开为wo 2019/215500)中,这些在先申请中的每一者的相关披露内容通过援引并入本文用于本文所引用的目的和主题。对于本文提供的任何grna序列,未明确指示修饰的那些意在包括未修饰的序列和具有任何合适的修饰的序列。
[0079]
本文披露的任何grna中的间隔子序列的长度可以取决于crispr/cas9系统和用于编辑本文披露的任何靶基因的组分。例如,来自不同细菌物种的不同cas9蛋白具有不同的最佳间隔子序列长度。因此,间隔子序列的长度可以具有5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、35、40、45、50、或超过50个的核苷酸。在一些实施例中,间隔子序列的长度可以具有18-24个核苷酸。在一些实施例中,靶向序列的长度可以具有19-21个核苷酸。在一些实施例中,间隔子序列的长度可包含20个核苷酸。
[0080]
在一些实施例中,grna可以是sgrna,其可以在sgrna序列的5’末端包含20个核苷酸间隔子序列。在一些实施例中,sgrna可以在sgrna序列的5’末端包含少于20个核苷酸的间隔子序列。在一些实施例中,sgrna可以在sgrna序列的5’末端包含大于20个核苷酸的间隔子序列。在一些实施例中,sgrna在sgrna序列的5’末端包含具有17-30个核苷酸的可变长度的间隔子序列。实例在实例5中的表8中提供。
[0081]
在一些实施例中,sgrna在sgrna序列的3’末端不包含尿嘧啶。在其他实施例中,sgrna可以在sgrna序列的3’末端包含一个或多个尿嘧啶。例如,sgrna可以在sgrna序列的3’末端包含1-8个尿嘧啶残基,例如,在sgrna序列的3’末端包含1、2、3、4、5、6、7或8个尿嘧啶残基。
[0082]
本文披露的任何grna,包括任何sgrna,可以是未修饰的。替代性地,它可以包含一个或多个经修饰的核苷酸和/或经修饰的主链。例如,经修饰的grna(例如sgrna)可以包含一个或多个2'-o-甲基硫代磷酸酯核苷酸,其可以位于5’末端、3’末端或这两个末端。
[0083]
在某些实施例中,一种以上的引导rna可以与crispr/cas核酸酶系统一起使用。每种引导rna可包含不同的靶向序列,以使crispr/cas系统切割一种以上的靶核酸。在一些实施例中,一种或多种引导rna可以在cas9 rnp复合物中具有相同或不同的性质,例如活性或稳定性。当使用一种以上引导rna时,每种引导rna可以在相同或不同的载体上编码。用于驱动一种以上引导rna表达的启动子是相同或不同的。
[0084]
应当理解,在本文所述的方法中可以使用一种以上的合适的cas9和一种以上的合适的grna,例如,本领域已知的或本文披露的那些。在一些实施例中,方法包括本领域已知的cas9酶和/或grna。实例可以在例如2018年5月11日提交的pct/ib 2018/001619(已公开为wo 2019/097305 a2)和2019年5月10日提交的pct/ib 2019/000500(现已公开为wo 2019/215500)中找到,这些在先申请中的每一者的相关披露内容通过援引并入本文用于本文所引用的目的和主题。
[0085]
cd70、trac和β2m基因的基因编辑
[0086]
在一些实施例中,如本文披露的富集的t细胞可以在本文披露的条件下经由crispr-cas9介导的基因编辑进行cd70基因、trac基因和β2m基因的基因编辑,与常规条件提供的那些相比,这将产生更高和更一致的基因编辑效率和更低的易位率。
[0087]
在具体实例中,靶向cd70基因的rnp复合物可包含约0.15mg/ml cas9(例如,seq id no:1的cas9)和约0.16mg/ml靶向cd70基因的grna(例如,cd70-7的grna)。rnp对基因编辑很有用,至少因为它们使在富含核酸的细胞环境中发生混杂相互作用的风险降到最低,并使rna免于降解。形成rnp的方法是本领域已知的。
[0088]
可以通过以下方式将本文披露的靶向cd70的rnp引入到富集的t细胞中:将rnp与合适量的富集的t细胞混合,并且在允许将rnp递送至细胞中的合适条件下对由此形成的混合物进行电穿孔。在一些情况下,富集的t细胞的合适量可在约100x106个细胞/ml至约400x106个细胞/ml的范围内。例如,用于第一电穿孔步骤的t细胞的合适量可在约200x106个细胞/ml至约350x106个细胞/ml的范围内。在一些实施例中,富集的t细胞的浓度可为约100x106个细胞/ml。在一些实施例中,富集的t细胞的浓度可为约200x106个细胞/ml。在一些实施例中,富集的t细胞的浓度可为约300x106个细胞/ml或约350x106个细胞/ml。
[0089]
电穿孔后,可以将具有破坏的cd70基因的t细胞在新鲜培养基中培养一段合适的时间以便恢复。基因编辑效率可以按照常规实践进行。可以对由此产生的经基因编辑的t细胞进行提高下游基因编辑效率的t细胞激活步骤及t细胞扩增步骤。
[0090]
trac基因编码tcr复合物的组分。trac基因的破坏导致tcr功能的丧失,并使工程化t细胞无同种异体反应性并适合同种异体移植,从而最小化移植物抗宿主病的风险。β2m基因编码主要组织相容性复合物(mhc)i复合物的公共(不变)组分。破坏β2m基因可防止宿主抗治疗性同种异体t细胞反应。敲除trac基因和β2m基因将使得产生用于细胞疗法的同种异体t细胞。
[0091]
在一些实施例中,本文披露的生产方法可以包括多个基因编辑步骤,以顺序编辑t细胞中的靶基因(cd70、trac和β2m)并将car编码核酸引入到t细胞中用于表达。每个基因编辑步骤可以涉及用于将引导rna、cas9酶和/或car编码核酸引入到t细胞中的电穿孔步骤以便对靶基因(cd70、trac和β2m)进行基因编辑并在t细胞中表达car。
[0092]
在一些实施例中,在第一电穿孔事件中编辑cd70,并且在第二电穿孔事件中编辑β2m/trac。参见例如图3a。然而,并不旨在将本文所述的方法限于该步骤顺序。图2a和图2b中提供的数据表明,在第一次电穿孔中递送的cd70和β2m的引导物均有利地使得易位率更低。因此,在其他实施例中,cd70和β2m均可以在第一电穿孔事件中被靶向。
[0093]
在一些情况下,可在第一电穿孔步骤中将靶向cd70基因的一种或多种引导rna和cas9酶引入到t细胞中以破坏cd70基因,并且可在第一电穿孔步骤之后的第二电穿孔步骤中将靶向trac和β2m基因的一种或多种引导rna、cas9酶及car编码核酸引入到t细胞中以破坏trac和β2m基因并将car编码核酸引入到t细胞中。在一些实例中,可在第1电穿孔步骤之后且在第2电穿孔步骤之前使用一种或多种t细胞激活剂(例如本文所述的那些)使t细胞激活。如以下实例3中所示,该设计允许在第二电穿孔步骤中对至少β2m基因的有效基因编辑,同时使具有由第一电穿孔步骤引起的破坏的cd70基因的t细胞保持高水平。
[0094]
在第一基因编辑步骤中,将包含第一cas9酶和靶向cd70基因的第一grna的第一rnp复合物引入到富集的t细胞中以产生具有破坏的cd70基因的t细胞。可以在进行第二基因编辑步骤之前激活此类t细胞以减少由第一基因编辑步骤导致的细胞损失。
[0095]
在第二基因编辑步骤中,将包含第二cas9酶和靶向trac基因的第二grna的第二rnp复合物以及包含第三cas9酶和靶向β2m基因的第三grna的第三rnp复合物引入到t细胞
中以产生具有破坏的cd70、trac、β2m基因的t细胞。cas9酶及靶向trac基因和β2m基因的grna可以形成一种或多种核糖核蛋白(rnp)复合物,可以将这些rnp复合物递送到具有如本文披露的破坏的cd70基因的激活的t细胞中。
[0096]
在一些实施例中,引入到具有破坏的cd70基因的t细胞(这些t细胞可任选地被激活)中的第二rnp复合物和第三rnp复合物可以含有相同量的cas9酶。例如,第二rnp复合物和第三rnp复合物均可包含约0.1至0.3mg/ml(例如,约0.1至0.2mg/ml)的cas9酶(例如,seq id no:1的cas9酶)。在一些实例中,第二rnp复合物和第三rnp复合物中的每一者可包含约0.15mg/ml的cas9酶,该cas9酶可以是seq id no:1的cas9酶。
[0097]
在其他实施例中,第二rnp复合物和第三rnp复合物可以含有不同量的cas9酶。在一些实例中,相对于靶向β2m基因的第三rnp复合物,靶向trac基因的第二rnp复合物可以包含更高量的cas9酶。替代性地,相对于靶向trac基因的第三rnp复合物,靶向β2m基因的第二rnp复合物可以包含更高量的cas9酶。
[0098]
第二rnp复合物和第三rnp复合物可以包含相同量的grna(一个靶向trac并且另一个靶向β2m)。替代性地,第二rnp复合物和第三rnp复合物可以包含不同量的grna。例如,靶向trac基因的grna的量可在约0.035mg/ml至约0.8mg/ml的范围内,例如约50μg/ml至约80μg/ml。在具体实例中,靶向trac基因的grna的量为约0.08mg/ml。替代性地或作为补充,靶向β2m基因的grna的量可在约0.075mg/ml至约0.3mg/ml的范围内,例如约0.1mg/ml至约0.3mg/ml。在具体实例中,靶向β2m基因的grna的量为约0.2mg/ml。
[0099]
在具体实例中,靶向trac基因的rnp复合物可包含约0.15mg/ml cas9(例如,seq id no:1的cas9)和约0.08mg/ml靶向trac基因的grna(例如,ta-1的grna)。替代性地或作为补充,靶向β2m基因的rnp复合物可包含约0.15mg/ml cas9(例如,seq id no:1的cas9)和约0.2mg/ml靶向β2m基因的grna(例如,β2m-1的grna)。
[0100]
在一些实施例中,可以将第二rnp复合物和第三rnp复合物依次通过电穿孔(即通过两个电穿孔事件)引入到激活的t细胞中。替代性地,可以将第二rnp复合物和第三rnp复合物同时(即通过一个电穿孔事件)引入到激活的t细胞中。在这种情况下,可以在电穿孔事件之前组合第二rnp复合物和第三rnp复合物以形成混合物。
[0101]
可以通过以下方式将本文披露的任何rnp引入到激活的t细胞中:将rnp与合适量的激活的t细胞混合,并且在允许将rnp递送至细胞中的合适条件下对由此形成的混合物进行电穿孔。在一些情况下,激活的t细胞的合适量可在约100x106个细胞/ml至约300x106个细胞/ml的范围内。例如,用于电穿孔步骤的t细胞的合适量可在约200x106个细胞/ml至约300x106个细胞/ml的范围内。在一些实例中,激活的t细胞的浓度可为约100x106个细胞/ml。在一些实施例中,激活的t细胞的浓度可为约200x106个细胞/ml。在一些实施例中,激活的t细胞的浓度可为约300x106个细胞/ml。
[0102]
在一些实施例中,激活的t细胞的合适量可在约1x108至约1x10
10
个细胞的范围内,例如约5x108至约8x109个细胞、约1x109至约5x109个细胞或约1x109至约3x109个细胞。
[0103]
用于电穿孔的t细胞可置于多个细胞盒中,这取决于所用的电穿孔仪器。合适的电穿孔仪器是本领域技术人员已知的,并且可以包括静态和流式电穿孔仪,包括lonza nucleofector、maxcyte gt和maxcyte gtx。在一些情况下,可以在电穿孔过程中使用多个细胞盒。更多细节提供于以下实例6中。
[0104]
在具体实例中,以上披露的第二rnp复合物和第三rnp复合物(包含总计约0.3mg/ml的cas9酶(例如,seq id no:1的cas9酶)、约0.08mg/ml的ta-1的grna和约0.2mg/ml的β2m-1的grna)可以与约100x106个细胞/ml至约400x106个细胞/ml(例如,约300x106个细胞/ml)的量的激活的t细胞混合。然后对混合物进行电穿孔以将rnp递送到t细胞中。
[0105]
在一些实例中,第一cas9酶、第二cas9酶和第三cas9酶是相同的,例如来自酿脓链球菌的cas9(spcas9)或包含seq id no:1的氨基酸序列的cas9酶。
[0106]
电穿孔后,可以将细胞在新鲜培养基中培养一段合适的时间以便恢复。基因编辑效率可以按照常规实践确定。可以对由此产生的经基因编辑的t细胞进行病毒载体转导以递送被配置用于car表达的核酸。
[0107]
(iii)t细胞激活
[0108]
可以对本文披露的任何t细胞(例如具有由第1电穿孔步骤引起的破坏的cd70基因的t细胞)进行激活步骤以允许t细胞增殖和t细胞扩增。本文披露的t细胞激活条件提供了高t细胞激活效率、高%car
+
表达并减少了由cd70基因的编辑导致的细胞损失。此外,与常规条件相比,本文披露的t细胞激活条件提供了更高的基因编辑效率和更大的编辑后t细胞扩增速率。参见以下实例。
[0109]
在一些实施例中,可使用t细胞激活剂(例如刺激cd3/tcr介导的信号传导途径和/或共刺激分子(例如cd28)介导的信号传导途径的试剂)实现t细胞激活。例如,t细胞激活剂可为cd3激动剂(例如激动性抗cd3抗体)并且激活cd3/tcr介导的细胞信号传导途径。替代性地或作为补充,t细胞激活剂可为cd28激动剂(例如抗cd28抗体)并且激活cd28介导的共刺激信号传导途径。在本文披露的方法中使用的任何t细胞激活剂可缀合至支持构件,例如纳米基质颗粒。在此类情况下,t细胞激活剂可缀合至相同的支持构件。替代性地,每种t细胞激活剂可缀合至不同的支持构件。在具体实例中,在本文披露的方法中使用的t细胞激活剂可包含抗cd3抗体和抗cd28抗体,这些抗体可缀合至纳米基质颗粒。在一些实施例中,t细胞激活剂包括附着于纳米基质颗粒的cd3激动剂和cd28激动剂。在一些实施例中,cd3激动剂和cd28激动剂附着于相同的纳米基质颗粒。在一些实施例中,cd3激动剂和cd28激动剂附着于不同的纳米基质颗粒。
[0110]
为了实现t细胞激活,可以将具有如本文披露的破坏的cd70基因的t细胞以合适的细胞接种密度和合适的细胞浓度置于细胞培养容器中,并在存在本文披露的任何t细胞激活剂的情况下温育一段合适的时间以诱导t细胞激活。
[0111]
在一些情况下,细胞培养容器中t细胞激活剂与细胞培养基的比率可在约1:10(v/v)至约1:15(v/v)的范围内。在一些实例中,细胞培养容器中t细胞激活剂与细胞培养基的比率可为约1:10(v/v)、约1:10.5(v/v)、约1:11(v/v)、约1:11.5(v/v)、约1:12(v/v)、约1:12.5(v/v)、约1:13(v/v)、约1:13.5(v/v)、约1:14(v/v)、约1:14.5(v/v)或约1:15(v/v)。在具体实例中,细胞培养容器中t细胞激活剂与培养基的比率为约1:12.5(v/v)。
[0112]
替代性地或作为补充,合适的细胞接种密度可为约1.0x106至2.5x106(例如,2x106/cm2)并且合适的细胞浓度可为约1.0x106至2.5x106(例如,2x106/ml)。可将具有破坏的cd70基因的t细胞与t细胞激活剂一起温育约60至80小时,例如约66小时或约72小时。
[0113]
替代性地或作为补充,合适的细胞接种密度可为约1.5x106至2.5x106(例如,2x106/cm2)并且合适的细胞浓度可为约1.5x106至2.5x106(例如,2x106/ml)。可将具有破坏
的cd70基因的t细胞与t细胞激活剂一起温育约66至80小时,例如约72小时。
[0114]
在一些实施例中,细胞培养容器可以是静态培养容器,这将允许如本文披露的基因工程化t细胞的相对大规模生产。与常规细胞培养瓶相比,静态细胞培养容器允许t细胞位于浸没在培养基下的高度透气的膜上,从而在不混合或摇动的情况下向t细胞供应氧气和营养物。静态培养容器允许在不更换培养基的情况下生产t细胞。因此,在一些实施例中,本文披露的任何方法中的t细胞激活过程可以不涉及培养基更换。
[0115]
当需要时,可以在下游基因编辑事件之前将激活剂从细胞培养容器中去除或稀释,以使激活剂在基因编辑期间可能带来的任何潜在影响最小化。在一些实施例中,可以使用常规方法例如离心从细胞培养容器中去除激活剂。替代性地,可以在基因编辑之前在细胞培养容器中稀释激活剂,例如通过向细胞培养容器中添加培养基来稀释。
[0116]
在一些实施例中,可以将来源于本文披露的任何t细胞激活过程的具有破坏的cd70基因的激活的t细胞培养过夜(例如,约16小时)以允许t细胞在基因编辑之前恢复。在一些情况下,具有破坏的cd70基因的激活的t细胞的培养物仍可含有t细胞激活剂。在其他情况下,具有破坏的cd70基因的激活的t细胞的培养物可具有很少或不存在t细胞激活剂。
[0117]
(iv)t细胞转导
[0118]
可以用包含编码嵌合抗原受体(car)的核酸序列的病毒载体例如腺相关病毒(aav)载体对具有敲除的cd70、trac和/或β2m基因的经基因编辑的t细胞进行转导以产生表达car的t细胞群体。
[0119]
嵌合抗原受体(car)
[0120]
嵌合抗原受体(car)是指人工免疫细胞受体,其经工程化以识别并结合不希望的细胞表达的抗原,例如疾病细胞表达的抗原,例如癌细胞表达的抗原。表达car多肽的t细胞被称为car t细胞。car具有以非mhc限制的方式将t细胞特异性和反应性重定向至所选靶标的能力。非mhc限制性抗原识别赋予car-t细胞识别独立于抗原加工的抗原的能力,从而绕开了肿瘤逃逸的主要机制。此外,当在t细胞上表达时,car不会有利地与内源性t细胞受体(tcr)α和β链二聚。
[0121]
有不同代的car,每一代都含有不同的组分。第一代car通过铰链结构域和跨膜结构域将抗体衍生的scfv与t细胞受体的cd3ζ(ζ或z)细胞内信号传导结构域连接。第二代car包含另外的共刺激结构域(例如,cd28、4-1bb(41bb)或icos)以提供共刺激信号。第三代car含有与tcr cd3ζ链融合的两个共刺激结构域(例如,cd27、cd28、4-1bb、icos或ox40的组合)。maude等人,blood.[血液]2015;125(26):4017-4023;kakarla和gottschalk,cancer j.[癌症杂志]2014;20(2):151-155)。不同代car构建体中的任何一代都在本披露的范围内。
[0122]
一般地说,car是融合多肽,其包含识别靶抗原的胞外结构域(例如,抗体的单链可变片段(scfv)或其他抗体片段)和胞内结构域,该胞内结构域包含t细胞受体(tcr)复合物的信号传导结构域(例如,cd3ζ),并且在大多数情况下包含共刺激结构域。(enblad等人,human gene therapy.[人类基因治疗]2015;26(8):498-505)。car构建体可以进一步包含位于胞外结构域和胞内结构域之间的铰链和跨膜结构域,以及用于表面表达的n末端的信号肽。信号肽的实例包括mlllvtslllcelphpafllip(seq id no:52)和malpvtalllplalllhaarp(seq id no:53)。可以使用其他信号肽。
[0123]
(a)抗原结合胞外结构域
[0124]
抗原结合胞外结构域是当car在细胞表面表达时暴露于胞外流体的car多肽的区域。在一些情况下,信号肽可以位于n末端,以促进细胞表面表达。在一些实施例中,抗原结合结构域可以是单链可变片段(scfv,其可以包括抗体重链可变区(vh)和抗体轻链可变区(v
l
)(以任一取向))。在一些情况下,vh和v
l
片段可通过肽接头连接。在一些实施例中,接头包括亲水性残基,即,多段甘氨酸和丝氨酸用于柔性,以及多段谷氨酸和赖氨酸用于增加溶解度。scfv片段保留了scfv片段衍生自的亲本抗体的抗原结合特异性。在一些实施例中,scfv可包含人源化vh和/或v
l
结构域。在其他实施例中,scfv的vh和/或v
l
结构域完全是人的。
[0125]
胞外抗原结合结构域可以对目标靶抗原,例如病理性抗原诸如肿瘤抗原有特异性。在一些实施例中,肿瘤抗原是“肿瘤相关抗原”,指免疫原性分子(例如蛋白),其通常在肿瘤细胞中的表达水平高于非肿瘤细胞中的表达水平,其在非肿瘤细胞可以不表达或仅在较低水平上表达。在一些实施例中,被携带肿瘤的宿主的免疫系统识别的肿瘤相关结构被称为肿瘤相关抗原。在一些实施例中,如果肿瘤相关抗原由大多数类型的肿瘤广泛表达,则其为通用肿瘤抗原。在一些实施例中,肿瘤相关抗原是分化抗原、突变抗原、过表达的细胞抗原或病毒抗原。在一些实施例中,肿瘤抗原是“肿瘤特异性抗原”或“tsa”,是指肿瘤细胞特有的免疫原性分子,例如蛋白。肿瘤特异性抗原仅在肿瘤细胞中表达,例如在特定类型的肿瘤细胞中表达。
[0126]
在一些实例中,本文披露的car构建体包含能够结合cd70的scfv胞外结构域。在一些实例中,本文披露的car构建体包含能够结合cd19的scfv胞外结构域。在一些实例中,本文披露的car构建体包含能够结合bcma的scfv胞外结构域。抗cd70 car的实例提供于以下实例中。
[0127]
(b)跨膜结构域
[0128]
本文披露的car多肽可以含有跨膜结构域,该跨膜结构域可以是跨膜的疏水性α螺旋。如本文所用,“跨膜结构域”是指在细胞膜、优选真核细胞膜中热力学稳定的任何蛋白结构。跨膜结构域可以提供含有其的car的稳定性。
[0129]
在一些实施例中,如本文提供的car的跨膜结构域可以是cd8跨膜结构域。在其他实施例中,跨膜结构域可以是cd28跨膜结构域。在又其他实施例中,跨膜结构域是cd8和cd28跨膜结构域的嵌合体。如本文提供的,可使用其他跨膜结构域。在一些实施例中,跨膜结构域是含有fvpvflpakptttpaprpptpaptiasqplslrpeacrpaaggavhtrgldfacdiyiwaplagtcgvlllslvitlycnhrnr(seq id no:54)或iyiwaplagtcgvlllslvitly(seq id no:55)的序列的cd8a跨膜结构域。可使用其他跨膜结构域。
[0130]
(c)铰链结构域
[0131]
在一些实施例中,铰链结构域可以位于car的胞外结构域(包含抗原结合结构域)与跨膜结构域之间或者car的胞质结构域与跨膜结构域之间。铰链结构域可以是起到将跨膜结构域连接至多肽链中的胞外结构域和/或胞质结构域的功能的任何寡肽或多肽。铰链结构域可以起到向car或其结构域提供柔性或防止car或其结构域的空间位阻的功能。
[0132]
在一些实施例中,铰链结构域可以包含多达300个氨基酸(例如,10至100个氨基酸或5至20个氨基酸)。在一些实施例中,一个或多个铰链结构域可以包括在car的其他区域
中。在一些实施例中,铰链结构域可以是cd8铰链结构域。可以使用其他铰链结构域。
[0133]
(d)细胞内信号传导结构域
[0134]
任何car构建体均含有一个或多个细胞内信号传导结构域(例如,cd3ζ,和任选地一个或多个共刺激结构域),其是受体的功能性末端。抗原识别后,受体聚簇,并且信号被传递到细胞。
[0135]
cd3ζ是t细胞受体复合物的细胞质信号传导结构域。cd3ζ包含三(3)个基于免疫受体酪氨酸的激活基序(itam),在t细胞与关联抗原接合后,它们将激活信号传输到t细胞。在许多情况下,cd3ζ提供主要的t细胞激活信号,但不提供完全感受态的激活信号,这需要共刺激信号传导。
[0136]
在一些实施例中,本文披露的car多肽可进一步包含一个或多个共刺激信号传导结构域。例如,cd28和/或4-1bb的共刺激结构域可用于传递完整的增殖/存活信号,以及cd3ζ介导的主要信号传导。在一些实例中,本文披露的car包含cd28共刺激分子。在其他实例中,本文披露的car包含4-1bb共刺激分子。在一些实施例中,car包括cd3ζ信号传导结构域和cd28共刺激结构域。在其他实施例中,car包括cd3ζ信号传导结构域和4-1bb共刺激结构域。在其他实施例中,car包括cd3ζ信号传导结构域、cd28共刺激结构域和4-1bb共刺激结构域。
[0137]
应当理解,本文描述的方法涵盖多于一种可用于产生表达car的基因工程化t细胞的合适的car,例如,本领域已知的或本文披露的那些。实例可以在例如2018年5月11日提交的pct/ib 2018/001619(已公开为wo 2019/097305 a2)和2019年5月10日提交的pct/ib 2019/000500中找到,这些在先申请中的每一者的相关披露内容通过援引并入本文用于本文所引用的目的和主题。
[0138]
例如,car结合cd70(也称为“cd70 car”或“抗cd70 car”)。结合cd70的示例性car的氨基酸序列在seq id no:46中提供(参见以下实例5中的表12)。
[0139]
用于将car构建体递送至t细胞的aav载体
[0140]
可以使用腺相关病毒(aav)将编码car构建体的核酸递送至细胞。aav是小病毒,其位点特异性整合到宿主基因组中,并且因此可以递送转基因,例如car。存在反向末端重复序列(itr),位于aav基因组和/或目的转基因的侧翼,并用作复制起点。aav基因组中还存在rep和cap蛋白,它们在转录时形成衣壳,这些衣壳封装aav基因组以递送到靶细胞中。这些衣壳上的表面受体会赋予aav血清型,其决定衣壳主要结合哪个靶器官,并且因此决定aav将最有效地感染哪些细胞。目前已知十二种人aav血清型。在一些实施例中,用于递送car编码核酸的aav是aav血清型6(aav6)。
[0141]
出于多种原因,腺相关病毒是用于基因疗法的最常用病毒之一。首先,aav在施用于包括人在内的哺乳动物后不会引起免疫反应。第二,将aav有效地递送至靶细胞,尤其是在考虑选择合适的aav血清型时。最后,因为基因组可以在宿主细胞中持续存在而不整合,aav具有感染分裂和非分裂细胞的能力。这种特性使它们成为基因疗法的理想候选。
[0142]
可以设计编码car的核酸,以插入宿主t细胞中的目标基因组位点中。在一些实施例中,靶基因组位点可以在安全港基因座中。
[0143]
在一些实施例中,编码car的核酸(例如,经由供体模板,其可由病毒载体诸如腺相关病毒(aav)载体携带)可被设计成使得其可插入trac基因内的位置以破坏基因工程化t细
胞中的trac基因并表达car多肽。trac的破坏导致内源性tcr的功能丧失。例如,trac基因中的破坏可以用核酸内切酶(如本文所述的那些)和靶向一个或多个trac基因组区域的一个或多个grnas来产生。对trac基因和靶区域特异的任何grna可用于此目的,例如,本文披露的那些。
[0144]
在一些实例中,trac基因中的基因组缺失和由car编码片段的替换可以通过同源定向修复或hdr(例如,使用供体模板,其可以是病毒载体例如腺相关病毒(aav)载体的一部分)来产生。在一些实例中,grna靶序列或其部分缺失(例如:seq id no:17)。在一些实施例中,trac基因中的破坏可以利用核酸内切酶(如本文所披露的那些)和靶向一个或多个trac基因组区域的一个或多个grna并将car编码片段插入trac基因中来产生。
[0145]
如本文披露的供体模板可以包含car的编码序列。在一些实例中,car编码序列的两侧可以有两个同源区,以允许使用crispr-cas9基因编辑技术在目的基因组位置处(例如,在trac基因处)的有效hdr。在这种情况下,靶基因座处的dna的两条链都可以被crispr cas9酶切割,该酶由靶位点特异性的grna引导。然后发生hdr,以修复双链断裂(dsb)并插入编码car的供体dna。为了使此正确发生,设计供体序列的侧翼残基与靶基因(例如trac基因)中dsb位点周围的序列(以下简称“同源臂”)互补。这些同源臂用作dsb修复的模板,并使hdr成为基本无错误的机制。同源定向修复(hdr)的速率是突变与切割位点之间的距离的函数,因此选择重叠或附近的靶位点很重要。模板可以包括同源区侧翼的额外序列或者可以含有与基因组序列不同的序列,从而可以进行序列编辑。
[0146]
替代性地,供体模板可以与dna中的靶位置不具有同源区域,并且可以通过在靶位点切割后通过nhej依赖性末端连接而整合。
[0147]
供体模板可以是单链和/或双链的dna或rna,并且可以线性或环状形式引入细胞中。如果以线性形式引入,则可以通过本领域技术人员已知的方法保护供体序列的末端(例如,以防止核酸外切降解)。例如,将一个或多个双脱氧核苷酸残基添加至线性分子的3'末端和/或将自身互补的寡核苷酸连接至一个或两个末端。参见,例如,chang等人,(1987)proc.natl.acad.sci.usa[美国国家科学院院刊]84:4959-4963;nehls等人,(1996)science[科学]272:886-889。保护外源多核苷酸免于降解的其他方法包括但不限于一个或多个末端氨基基团的添加和经修饰的核苷酸间键的使用,例如,硫代磷酸酯、氨基磷酸酯和o-甲基核糖或脱氧核糖残基。
[0148]
可以将供体模板作为载体分子的一部分引入细胞中,该载体分子具有另外的序列,诸如例如,复制起点、启动子和编码抗生素抗性的基因。此外,供体模板可以作为裸露的核酸引入细胞(作为与试剂(例如脂质体或泊洛沙姆)复合的核酸引入),或可以通过病毒递送(例如,腺病毒、aav、疱疹病毒、逆转录病毒、慢病毒和整合酶缺陷型慢病毒(idlv))。
[0149]
在一些实施例中,供体模板可以插入在内源启动子附近的位点(例如,下游或上游),从而其表达可以由内源启动子驱动。在其他实施例中,供体模板可包含外源启动子和/或增强子,例如组成型启动子、诱导型启动子或组织特异性启动子,以控制car基因的表达。在一些实施例中,外源启动子是ef1α启动子。可以使用其他启动子。
[0150]
此外,外源序列还可包括转录或翻译调控序列,例如,启动子、增强子、绝缘子、内部核糖体进入位点、编码2a肽的序列和/或聚腺苷酸化信号。
[0151]
t细胞转导
[0152]
可以将合适量的编码本文披露的car构建体(例如抗cd70 car)的任何病毒载体(例如aav载体)与合适量的t细胞(例如本文披露的经基因编辑的t细胞)一起温育一段合适的时间以允许病毒载体进入t细胞。例如,转导过程可以涉及使用一定范围的优化的感染复数(moi),从而增加car
+
t细胞的百分比。在一些情况下,转导过程中aav载体的moi可在约1,000至约150,000的范围内,例如约10,000至约80,000。在一些实例中,转导过程中使用的aav载体的moi可为约1,000至约150,000、约5,000至约100,000、约10,000至约100,000、约10,000至约90,000、约10,000至约80,000、约10,000至约70,000、约10,000至约60,000、约10,000至约50,000、约10,000至约40,000、约10,000至约30,000、约10,000至约20,000、约20,000至约80,000、约30,000至约80,000、约40,000至约80,000、约50,000至约80,000、约60,000至约80,000、或约70,000至约80,000。在一些实例中,转导过程中使用的aav载体的moi可为约1,000、约2,500、约5,000、约10,000、约15,000、约20,000、约25,000、约30,000、约31,000、约32,000、约33,000、约34000、约35,000、约40,000、约50,000、约60,000、约70,000、约80,000、约90,000、约100,000、约110,000、约120,000、约130,000、约140,000或约150,000。
[0153]
在一些实施例中,aav载体编码抗cd70 car(例如,如以下实例5中的表12中所披露),并且用于转导过程的这种aav载体的moi为约20,000。在其他实施例中,aav载体编码抗cd19 car,并且用于转导过程的这种aav载体的moi为约20,000。在其他实施例中,aav载体编码抗bcmacar,并且用于转导过程的这种aav载体的moi为约20,000。
[0154]
转导后,可以将t细胞在合适的细胞培养基中培养一段合适的时间以便恢复。具有敲除的cd70、trac和β2m基因并表达car的基因工程化t细胞可以如下文披露的那样在体外扩增。
[0155]
(v)t细胞扩增
[0156]
本文披露的基因工程化t细胞可以在合适的条件下在体外扩增以产生达到临床相关规模的基因工程化t细胞群体。在该扩增步骤中使用的细胞培养条件至少部分地旨在在较短的温育时间段内实现较高的最终细胞密度(从而降低生产成本)和用于细胞疗法的较高效力的t细胞。效力可以通过各种t细胞功能来指示,例如增殖、靶细胞杀伤、细胞因子产生、激活、迁移以及它们的组合。
[0157]
在一些实施例中,可以通过将t细胞群体(例如,本文披露的基因工程化t细胞)以细胞容器中约150,000个细胞/cm2至约600,000个细胞/cm2的接种密度接种于细胞培养容器中来进行t细胞扩增步骤。例如,可以将t细胞以约300,000个细胞/cm2至约500,000个细胞/cm2接种于细胞容器中。在一些方面,通过将t细胞群体以至少约60,000个细胞/cm2、至少约62,500个细胞/cm2或至少约83,000个细胞/cm2的接种密度接种于细胞培养容器中来进行t细胞扩增。在一些方面,通过将t细胞群体以至少约150,000个细胞/cm2或至少约250,000个细胞/cm2或至少约300,000个细胞/cm2或至少约400,000个细胞/cm2或至少约500,000个细胞/cm2或至少约600,000个细胞/cm2的接种密度接种于细胞培养容器中来进行t细胞扩增。在一些方面,接种密度为约250,000个细胞/cm2。在其他方面,接种密度为约500,000个细胞/cm2。在其他方面,接种密度为约600,000个细胞/cm2。
[0158]
在一些实施例中,可以通过以下方式进行t细胞扩增步骤:将t细胞群体(例如,本文披露的基因工程化t细胞)以约2x105个细胞/cm2至约7x105个细胞/cm2的接种密度接种于
细胞培养容器中,并且将细胞培养约6天至约12天。在一些实例中,通过以下方式进行t细胞扩增:将t细胞群体以约2x105个细胞/cm2至约7x105个细胞/cm2、约2x105个细胞/cm2至约5x105个细胞/cm2、约2x105个细胞/cm2至约4x105个细胞/cm2、2x105个细胞/cm2至约3x105个细胞/cm2、3x105个细胞/cm2至约5x105个细胞/cm2、或4x105个细胞/cm2至约5x105个细胞/cm2的接种密度接种于细胞培养容器中,并且将细胞培养约6天至约12天、约6天至约11天、约6天至约10天、约6天至约9天、约6天至约8天、约6天至约7天、约7天至约12天、约7天至约11天、约7天至约10天、约7天至约9天、约7天至约8天、约8天至约12天、约8天至约9天、约9天至约12天、约10天至约12天、或约11天至约12天。在一些实施例中,通过以下方式进行t细胞扩增:将t细胞群体以约3x105个细胞/cm2至约5x105个细胞/cm2的接种密度接种于细胞培养容器中,并且将细胞培养约7天至约9天。
[0159]
在一些实施例中,t细胞扩增步骤可包括重新铺板细胞培养物(即,将细胞培养物分成新的培养容器)。在一些实施例中,可以在编辑后第3、4、5、6或7天以1:4比率(1个容器分成4个新容器)重新铺板细胞培养物以便进一步扩增。
[0160]
t细胞扩增可以在静态培养容器中进行,这允许在不更换培养基的情况下扩增t细胞。例如,可以在不更换培养基的情况下将t细胞在静态培养容器中扩增约7天至约12天或约7天至约9天。
[0161]
(vi)tcrαβ
+
t细胞的耗尽
[0162]
在一些实施例中,可以从本文披露的扩增的t细胞群体中耗尽tcrαβ
+
t细胞,以产生用于细胞疗法的同种异体t细胞群体。如本文所用,“tcrαβ
+
t细胞耗尽”是指从包含这种细胞的细胞群体中耗尽tcrαβ
+
t细胞。在tcrαβ
+
t细胞耗尽后,所得t细胞群体可具有实质上低水平的tcrαβ
+
t细胞(例如,总细胞群体中小于3%,或总细胞群体中小于2%、小于1%或小于0.5%)。在一些实例中,所得t细胞群体可不含tcrαβ
+
t细胞,即tcrαβ
+
t细胞的存在不可通过常规方法(例如,在使用与tcrαβ
+
结合的抗体的免疫测定法中或通过流式细胞术)检测。
[0163]
可以通过以下方式进行tcrαβ
+
t细胞耗尽:使用识别tcrαβ
+
t细胞的试剂捕获tcrαβ
+
t细胞,从而将它们与缺乏tcrαβ
+
的那些细胞分离,例如通过进行磁性细胞分离。可以通过将上文披露的扩增的t细胞与其上固定有抗tcrαβ抗体的珠接触并收集未结合的细胞来进行这些方法。可培养由此收集的未结合的细胞(缺乏tcrαβ
+
的那些细胞)以允许细胞预先恢复,例如,可将未结合的细胞培养过夜以允许细胞恢复。
[0164]
(vii)基因工程化t细胞的收获
[0165]
然后可以使用本领域已知的常规方法收获通过本文披露的任何方法产生的基因工程化t细胞以用于治疗用途。例如,收获基因工程化t细胞可包括收集已耗尽tcrαβ
+
的细胞。收获的基因工程化t细胞群体可用作药物物质。如本文所用,“药物物质”是指可施用给患者的基因工程化t细胞群体。可以将药物物质配制用于治疗用途,例如,在储存培养基(例如,cryostor cs5)中配制并冻存以备将来使用。
[0166]
可以测试药物物质的一种或多种污染物,例如支原体、人类病毒(例如hiv、hbv、hcv、cmv)和细菌内毒素。替代性地或作为补充,可以测试药物物质的无菌性。可以将无污染的药物物质等分成单独患者剂量。替代性地或作为补充,可以储存无污染的药物物质以用于治疗用途。
[0167]
因此,本披露的各方面提供了基因工程化t细胞群体(药物物质)。基因工程化t细胞群体具有破坏的cd70基因、破坏的trac基因、破坏的β2m基因和编码car的核酸,例如本文所述的那些。在一些实施例中,car结合在病理细胞上表达的抗原。在一些实施例中,car结合cd70。在一些实施例中,car结合cd19。在一些实施例中,car结合bcma。
[0168]
在一些实施例中,通过本文所述方法产生的至少30%、至少40%、至少50%、至少60%、至少70%、至少80%、至少90%或至少95%的基因工程化t细胞群体表达car。在其他方面,表达car的这些细胞还不表达可检测水平的表面cd70和/或可检测水平的表面tcr和/或可检测水平的表面β2m。
[0169]
在其他实施例中,在通过本文所述方法产生的至少30%的基因工程化t细胞群体表达car的情况下,该细胞群体包含不超过约5%、不超过约2%或不超过约1%的表达表面cd70的t细胞。
[0170]
在其他实施例中,在通过本文所述方法产生的至少30%的基因工程化t细胞群体表达car的情况下,该细胞群体包含不超过约1.0%、不超过约0.5%、不超过约0.4%或不超过约0.15%的表达表面tcr的t细胞(例如,tcrα/β+细胞)。
[0171]
在其他实施例中,在通过本文所述方法产生的至少30%的基因工程化t细胞群体表达car的情况下,该细胞群体包含不超过约50%、不超过约40%或不超过约30%的表达表面β2m的t细胞。
[0172]
通过本文所述方法产生的基因工程化t细胞群体也在本披露的范围内,该基因工程化t细胞群体包含cas9酶、靶向cd70基因的grna、靶向trac基因的grna、靶向β2m基因的grna和aav载体,该aav载体包含编码car(例如,cd70 car或cd19 car或bcma car)的核酸序列。
[0173]
ii.治疗应用
[0174]
可以将通过本文所述方法产生的基因工程化t细胞群体施用给受试者以用于治疗目的,例如治疗由基因工程化t细胞群体表达的car构建体靶向的癌症。
[0175]
受试者可以是期望对其进行诊断,治疗或疗法的任何受试者。在一些实施例中,该受试者是哺乳动物。在一些实施例中,该受试者是人。
[0176]
可使用通过本文所述方法产生的基因工程化t细胞群体治疗的癌症的非限制性实例包括但不限于多发性骨髓瘤、白血病(例如t细胞白血病、b细胞急性成淋巴细胞性白血病(b-all)和/或慢性淋巴细胞性白血病(c-cll))、淋巴瘤(例如b细胞非霍奇金淋巴瘤(b-nhl)、霍奇金淋巴瘤和/或t细胞淋巴瘤)和/或透明细胞肾细胞癌(ccrcc)、胰腺癌、胃癌、卵巢癌、宫颈癌、乳腺癌、肾癌、甲状腺癌、鼻咽癌、非小细胞肺癌(nsclc)、成胶质细胞瘤和/或黑素瘤。
[0177]
施用可包括通过引起基因工程化t细胞群体至少部分定位在所需位点(诸如肿瘤位点)的方法或途径将基因工程化t细胞群体置于(例如,移植到)受试者体内,使得可产生所需的效果。基因工程化t细胞群体可以通过任何适当的途径施用,该途径导致递送至受试者中的所需位置,在该位置中至少一部分植入的细胞或细胞组分保持活力。在施用于受试者后,细胞的活力期可以短至数小时(例如二十四小时)、几天,长达数年,或甚至受试者的寿命(即长期植入)。例如,在本文所述的一些方面,有效量的基因工程化t细胞群体可以经由全身性施用途径(如腹膜内或静脉内途径)施用。
[0178]
在一些实施例中,全身性施用基因工程化t细胞群体,这是指将细胞群体以不同于直接施用至靶部位、组织或器官的方式施用,而是使其进入受试者的循环系统,从而经受代谢和其他类似过程。施用的合适模式包括注射、输注、滴注或摄取。注射包括但不限于静脉内、肌内、动脉内、鞘内、心室内、囊内、眶内、心内、真皮内、腹膜内、经气管、皮下、表皮下、关节内、被膜下、蛛网膜下、脊柱内、脊髓内和胸骨内注射和输注。在一些实施例中,该途径是静脉内。
[0179]
有效量是指预防或减轻医学病症(例如,癌症)的至少一种或多种体征或症状所需的基因工程化t细胞群体的量,且涉及足以提供所希望的效果(例如,治疗患有医学病症的受试者)的基因工程化t细胞群体的量。有效量还包括足以预防或延迟疾病症状的发展、改变疾病症状的进程(例如但不限于减慢疾病症状的进展)或逆转疾病症状的量。应当理解,对于任何给定的情况,本领域的普通技术人员可以使用常规实验来确定适当的有效量。
[0180]
有效量的基因工程化t细胞群体可包括至少102个细胞、至少5x102个细胞、至少103个细胞、至少5x103个细胞、至少104个细胞、至少5x104个细胞、至少105个细胞、至少2x105个细胞、至少3x105个细胞、至少4x105个细胞、至少5x105个细胞、至少6x105个细胞、至少7x105个细胞、至少8x105个细胞、至少9x105个细胞、至少1x106个细胞、至少2x106个细胞、至少3x106个细胞、至少4x106个细胞、至少5x106个细胞、至少6x106个细胞、至少7x106个细胞、至少8x106个细胞、至少9x106个细胞或它们的倍数。
[0181]
使用如本文所述的那样生产的基因工程化t细胞群体的治疗功效可由本领域普通技术人员确定。如果疾病(例如,癌症)的任何一种或所有体征或症状(举一个例子,功能性靶标的水平)以有益的方式改变(例如,增加至少10%)或其他临床上可接受的症状或标志物得到改善或减轻,则该治疗被认为是“有效的”。功效还可以通过如通过住院治疗或需要医疗干预所评估的受试者恶化的失败(例如,疾病进展停止或至少减慢)来测量。测量这些指标的方法是本领域技术人员已知的和/或本文所述的。治疗包括对受试者疾病的任何治疗,包括:(1)抑制疾病,例如阻止或减缓症状的进展;或者(2)减轻疾病,例如引起症状消退;以及(3)预防或降低症状发展的可能性。
[0182]
如本文所述的那样生产的基因工程化t细胞群体也可用于组合疗法中。例如,如本文所述的那样生产的基因工程化t细胞群体可以与其他治疗剂共同使用,用于治疗相同指征,或用于增强基因工程化t细胞群体的功效和/或降低基因工程化t细胞群体的副作用。
[0183]
通用技术
[0184]
除非另有指示,否则本披露的实践将采用分子生物学(包括重组技术)、微生物学、细胞生物学、生物化学和免疫学的常规技术,这些技术在本领域的技术范围内。此类技术在文献中有充分说明,诸如molecular cloning:a laboratory manual[分子克隆:实验室手册],第二版(sambrook等人,1989)cold spring harbor press[冷泉港出版社];oligonucleotide synthesis[寡核苷酸合成](m.j.gait编辑,1984);methods in molecular biology[分子生物学方法],humana press[哈玛纳出版社];cell biology:a laboratory notebook[细胞生物学:实验室笔记本](j.e.cellis编辑,1989)academic press[学术出版社];animal cell culture[动物细胞培养](r.i.freshney编辑1987);introduction to cell and tissue culture[细胞和组织培养导论](j.p.mather和p.e.roberts,1998)plenum press[普莱纽姆出版社];cell and tissue culture:
laboratory procedures[细胞和组织培养:实验室操作](a.doyle、j.b.griffiths和d.g.newell编辑,1993-8)j.wiley and sons[威利父子出版社];methods in enzymology[酶学方法](academic press,inc.[学术出版社公司]);handbook of experimental immunology[实验免疫学手册](d.m.weir和c.c.blackwell编辑):gene transfer vectors for mammalian cells[哺乳动物细胞的基因转移载体](j.m.miller和m.p.calos编辑,1987);current protocols in molecular biology[最新分子生物学实验方法汇编](f.m.ausubel等人编辑1987);pcr:the polymerase chain reaction[pcr:聚合酶链反应](mullis等人编辑1994);current protocols in immunology[免疫学现行方案](j.e.coligan等人编辑,1991);short protocols in molecular biology[分子生物学简短方案](wiley and sons[威利父子出版社],1999);immunobiology[免疫生物学](c.a.janeway和p.travers,1997);antibodies[抗体](p.finch,1997);antibodies:a practice approach[抗体:实用方法](d.catty.编辑,irl press[irl出版社],1988-1989);monoclonal antibodies:a practical approach[单克隆抗体:实用方法](p.shepherd和c.dean编辑,oxford university press[牛津大学出版社],2000);using antibodies:laboratory manual[使用抗体:实验室手册](e.harlow和d.lane(cold spring harbor laboratory press[冷泉港实验室出版社],1999);the antibodies[抗体](m.zanetti和j.d.capra编辑harwood academic publishers[哈伍德学术出版社],1995);dna cloning:a practical approach[dna克隆:实用方法],第i卷和第ii卷(d.n.glover编辑1985);nucleic acid hybridization[核酸杂交](b.d.hames和s.j.higgins编辑1985);transcription and translation[转录和翻译](b.d.hames和s.j.higgins编辑1984);animal cell culture[动物细胞培养](r.i.freshney编辑1986);immobilized cells and enzymes[固定化细胞和酶](lrl press[lrl出版社],1986);以及b.perbal,a practical guide to molecular cloning[分子克隆实用指南](1984);f.m.ausubel等人(编辑)。
[0185]
无需进一步详细阐述,相信本领域的普通技术人员可以基于以上描述在其最大程度上利用本发明。因此以下特定实施例将被解释为仅是说明性的,并且无论如何并非以任何方式限制本披露的其余内容。本文引用的所有出版物均为本文引用的目的或主题通过援引并入。
[0186]
实例
[0187]
为了更充分地理解所描述的本发明,阐述以下实例。提供本技术中描述的实例以说明本文提供的方法和组合物,并且不应以任何方式解释为限制其范围。
[0188]
实例1:t细胞富集的优化条件的鉴定。
[0189]
本实例报告了使用自动化细胞处理系统从白细胞单采样富集cd4
+
和cd8
+
t细胞来进行t细胞富集的优化条件的鉴定。
[0190]
方法
[0191]
白细胞单采样和缓冲液制备
[0192]
从血液医疗公司(hemacare)或干细胞快运公司(stem express)收集人白细胞单采样并处理用于t细胞富集。pbs/edta缓冲液(磷酸盐缓冲盐水,ph 7.2,补充有1mm edta)补充有0.5%人血清白蛋白(hsa),并在t细胞选择期间用于处理、预激、洗涤和洗脱。
[0193]
对白细胞单采样供体进行以下筛查:
technologies),目录:amqax1000)进行细胞计数和活力评估。通过上下吸移几次将细胞(20μl)与台盼蓝(20μl)混合而不引入气泡。将细胞/台盼蓝混合物(10μl)加载到ii细胞计数腔室玻片中。
[0213]
流式细胞术
[0214]
在室温(rt)下,用95μl染色缓冲液(0.5%牛血清白蛋白(bsa)/dpbs)中的5μl人trustain fcx
tm
封闭约1x106个总核细胞10分钟。进一步将细胞与pacific blue缀合的抗人cd45抗体(1:50)、bv510缀合的抗人cd3抗体(1:50)、apc-cy7缀合的抗人cd4抗体(1:50)、pe-cy7缀合的抗人cd8抗体(1:50)、apc缀合的抗人cd19抗体(1:50)、fitc缀合的抗人cd56抗体(1:50)和pe缀合的抗人cd33抗体(1:50)一起在4℃下温育30分钟。然后,将含有5μl 7-氨基-放线菌素d(7-aad)活力染色溶液的1ml氯化铵钾(ack)裂解缓冲液施加于每个样品。在室温下与ack裂解缓冲液一起温育10分钟后,用novocyte-3000流式细胞仪采集细胞。
[0215]
结果
[0216]
白细胞单采样品中的白细胞(wbc)
[0217]
在测试的白细胞单采样中wbc在8.14x109至21.36x109个细胞的范围内,而淋巴细胞数量在5.77x109至17.32x109的范围内。
[0218]
cd4和cd8富集-纯度、活力、细胞恢复率和产率
[0219]
在测试的9个批次中,使用程序a评价四个批次并且使用程序b评价五个批次。所有批次产生具有》90%纯度和》90%活力的t细胞(表2)。程序a的细胞恢复率为31%,而程序b的细胞恢复率为55.69%。
[0220]
表2.cd4和cd8富集结果
[0221][0222]
总之,这些结果表明在来自健康供体(hd)白细胞单采样的t细胞中以高纯度(》90%)和高活力(》90%)富集了cd4
+
和cd8
+
t细胞。
[0223]
实例2:t细胞激活的优化条件的鉴定。
[0224]
本实例报告了使用缀合至重组人源化cd3和cd28激动剂的胶体聚合物纳米基质进行t细胞激活的优化条件的鉴定。对在不同条件下激活的t细胞检查基因编辑和/或car表达水平以鉴定实现优异基因编辑和/或car表达水平的优化t细胞激活条件。简言之,以小规模过程生产基因工程化t细胞,在该过程中将富集的t细胞解冻,随后在激活前用一次电穿孔或两次电穿孔激活48小时或72小时,并且在转导后7天通过流式细胞术确定%car
+
表达。
[0225]
为了开始该小规模生产过程,从液氮贮罐中取出冷冻管,并在水浴中解冻,直到剩余少量冷冻材料。然后将细胞滴加到10x体积的完全生长培养基(x-vivo
tm 15,5%人ab血清,50ng/ml il7和10ng/ml il2)中,并通过在室温下以300g离心10分钟沉淀。将细胞重悬达到1x106个细胞/ml的浓度并且经受与胶体聚合物纳米基质缀合的重组人源化cd3和cd28激动剂介导的激活,从而改善下游修饰,或对细胞进行电穿孔以引入用于crispr-cas9依赖性基因编辑的组分。
[0226]
用与胶体聚合物纳米基质共价连接的重组cd3和cd28激活分离的t细胞。将缀合至重组人源化cd3和cd28激动剂的胶体聚合物纳米基质以1:12.5比率或40μl/1x106个细胞施加于未处理的瓶中的细胞。将细胞与缀合至重组人源化cd3和cd28激动剂的胶体聚合物纳米基质一起在培养箱中在37℃、5%co2下保持48小时或72小时。温育后,将细胞在室温下以300g离心10分钟。然后将细胞沉淀物重悬于完全生长培养基中,并在基因修饰前以1x106个细胞/ml的浓度培养过夜。
[0227]
对于电穿孔,通过添加台盼蓝并在细胞计数器上计数来定量总细胞数和细胞活力。然后,将细胞在室温下以300g离心10分钟。在10ml电穿孔缓冲液中洗涤细胞沉淀物并再次离心。在离心细胞的同时,制备核糖核蛋白(rnp)复合物。如果进行多次编辑,则单独形成rnp复合物,然后组合在一起。使用指示浓度的grna和cas9形成四种单独的rnp复合物(表3)。每种rnp复合物使用包含seq id no:1的cas9形成。cas9和grna序列也参见实例5。
[0228]
表3.含有grna和cas9的rnp复合物。
[0229][0230]
使用基于流式电穿孔的转染系统对细胞进行电穿孔。一旦对每个单独的比色皿进行电穿孔,就将细胞和rnp溶液等分到未处理的12孔板中,每个孔含有500μl x-vivo
tm 15培养基(不含人ab血清、il2或il7)。使细胞在培养箱中静置20分钟。通过添加台盼蓝并在细胞计数器上计数来定量总细胞数和细胞活力。
[0231]
基于静置后的总细胞数,可能需要用x-vivo
tm 15(不含人ab血清、il2或il7)进一步稀释细胞以达到所需浓度。需要总细胞数来计算进行转导所需的aav体积。
[0232]
所需aav的μl=(总细胞数)(期望moi(即,20k))/(病毒vgc/ml(即,1.5x10
13
))
[0233]
将aav和细胞悬液混合并使之在未处理的瓶中在37℃和5%co2下温育1小时。将包含aav的全部体积添加到含有100ml完全培养基的静态培养容器中。将静态培养容器温育3天以允许细胞扩增。
[0234]
电穿孔后,用100ml完全生长培养基填充静态培养容器的每个孔。将经基因修饰的细胞以5x105个细胞/ml至1x106个细胞/ml的浓度接种于100ml的完全生长培养基中。将静态培养容器温育三至四天以允许细胞扩增。每三至四天补充il2和il7达到100u/ml或10ng/ml il2和50ng/ml il7的最终工作浓度。每三至四天通过添加台盼蓝并在细胞计数器上计数来定量总细胞数。电穿孔后将细胞保持培养九至十二天以实现最大的总细胞数。
[0235]
(i)t细胞激活的优化条件增加了%car
+
表达
[0236]
使用电穿孔将grna和cas9引入到t细胞中,以对包括cd70、pd1、β2m和trac基因的四种靶基因进行crispr-cas9依赖性基因编辑。进行单次电穿孔以同时靶向所有四种基因。当进行两次电穿孔时,在第一次电穿孔中将靶向cd70和pd1基因的rnp复合物引入到t细胞中,并且在第二次电穿孔中将靶向β2m和trac基因的rnp复合物引入到那些t细胞中。
[0237]
如表4所示,在一次电穿孔或两次电穿孔之前激活48小时的t细胞分别显示出54.7%和57.5%的%car
+
表达。激活72小时的t细胞表现出比激活48小时的t细胞多大约10%的总%car
+
表达,无论对t细胞进行一次还是两次电穿孔(表4)。
[0238]
表4.激活48小时或72小时的t细胞的%car
+
表达。
[0239][0240]
这些结果表明,与48小时的t细胞激活所提供的相比,72小时的t细胞激活增加了%car
+
表达。当靶向pd1的rnp复合物不包括在电穿孔中时,观察到类似的结果。
[0241]
(ii)t细胞激活的优化条件减少了电穿孔的细胞损失
[0242]
对t细胞进行第一电穿孔步骤以引入用于cd70基因和pd1基因的crispr-cas9依赖性编辑的组分。在t细胞激活48小时或72小时之前和之后确定细胞数。
[0243]
如表5所示,当t细胞激活48小时时,第二次电穿孔之前获得的细胞计数小于最初接种用于激活的细胞数。相反,当t细胞激活72小时时,第二次电穿孔之前获得的细胞计数大于最初接种用于激活的细胞数(表5)。
[0244]
表5.在t细胞激活48小时或72小时之前和之后的细胞数。
[0245]
[0246]
这些结果表明t细胞激活72小时减少了t细胞仅激活48小时时观察到的第一次电穿孔后的细胞损失。当靶向pd1的rnp复合物不包括在电穿孔中时,观察到类似的结果。
[0247]
实例3:β2m敲除的优化条件的鉴定。
[0248]
本实例报告了使用crispr-cas9依赖性基因编辑敲除β2m的优化条件的鉴定。β2m的敲除可以在第一次电穿孔或第二次电穿孔中进行。tcr的敲除通常在第二次电穿孔中或转导之前进行,以确保hdr介导的cd70 car的插入。cd70的敲除通常在初始电穿孔中进行,以防止在插入cd70 car之前可能的细胞间相互杀伤。
[0249]
简言之,在小规模过程中生产基因工程化t细胞,在该过程中形成靶向β2m的rnp复合物,并经由单次电穿孔或两步电穿孔过程将其引入到t细胞中。详见以上实例2。
[0250]
为了在第一次电穿孔中敲除β2m,在第一次电穿孔中将靶向cd70和β2m基因的rnp复合物引入到t细胞中,并且在第二次电穿孔中将靶向pd1和trac基因的rnp复合物引入到t细胞中。为了在第二次电穿孔中敲除β2m,在第一次电穿孔中将靶向cd70和pd1基因的rnp复合物引入到t细胞中,并且在第二次电穿孔中将靶向β2m和trac基因的rnp复合物引入到t细胞中。还在单个电穿孔事件中用靶向cd70、pd1、β2m和trac基因的rnp复合物对t细胞进行电穿孔。
[0251]
如表6所示,当靶向β2m的rnp复合物包括在第一次电穿孔中时,在转导后7天残余β2m
+
表达为约60%,无论t细胞激活48小时还是72小时。当靶向β2m的rnp复合物包括在单次电穿孔或第二次电穿孔中时,残余β2m
+
表达为约20%(表6)。在转导后7天检测不到残余cd70
+
表达(表7)。残余cd70
+
表达细胞可能已通过用靶向cd70的rnp复合物敲除而消除,或通过cd70 car
+
细胞消除。对于所测试的每种β2m敲除条件,观察到类似的t细胞生长和t细胞活力(图1)。
[0252]
表6.β2m敲除条件对β2m表达的影响。
[0253][0254]
表7.β2m敲除条件对cd70表达的影响。
[0255][0256]
这些结果表明,在第二电穿孔步骤中引入靶向β2m的rnp复合物提供了β2m的优异敲除,同时保持cd70的有效敲除或细胞生长和细胞活力。当靶向pd1的rnp复合物不包括在电穿孔中时,观察到类似的结果。
[0257]
实例4:t细胞电穿孔的优化条件的鉴定。
[0258]
该实例报告了通过电穿孔将crispr-cas9依赖性基因编辑的多种rnp复合物引入到t细胞中的优化条件的鉴定。
[0259]
简言之,在小规模过程中生产基因工程化t细胞,在该过程中经由单次电穿孔或两步电穿孔过程将rnp复合物引入到t细胞中。详见以上实例2。通过ddpcr确定易位率。
[0260]
除了当靶向pd1和cd70的rnp复合物在第一次电穿孔中组合在一起时之外,用一次电穿孔进行基因工程化的t细胞显示出比在两步中电穿孔的t细胞显著更高的易位率(图2a)。当在第一次电穿孔中(通过rnp复合物)递送靶向cd70的grna时,易位率小于2%。参见图2a和图2b。用四种rnp复合物一起电穿孔的t细胞的细胞遗传学分析揭示了易位可能发生在容纳pd1(第2号染色体)、β2m(第15号染色体)、tcr(第14号染色体)和cd70(第19号染色体)的染色体中(数据未示出)。
[0261]
总之,这些结果表明,可以通过以两步进行的电穿孔引入rnp复合物来实现较低的易位率。当靶向pd1的rnp复合物不包括在电穿孔中时,观察到类似的结果。
[0262]
实例5:用于制备表达抗cd70 car并具有基因破坏的cd70、trac和β2m基因的基因工程化t细胞(ctx130)的生产过程开发。
[0263]
概述
[0264]
ctx130是一种cd70定向的t细胞免疫疗法,其由使用crispr/cas9(规律间隔成簇短回文重复序列/crispr相关蛋白9)基因编辑组分(sgrna和cas9核酸酶)离体基因修饰的同种异体t细胞构成。
[0265]
这些修饰包括t细胞受体α恒定区(trac)、β2m和cd70的靶向破坏。trac基因座的破坏导致t细胞受体(tcr)表达丧失,目的是降低移植物抗宿主病(gvhd)的概率,而β2m基因座的破坏导致主要组织相容性复合物i型(mhc i)蛋白表达缺失,目的是通过降低宿主排斥的概率来改善持久性。cd70的破坏导致cd70表达丧失,从而防止在插入cd70 car之前可能的细胞间相互杀伤。添加抗cd70 car将修饰的t细胞定向至表达cd70的肿瘤细胞。
[0266]
抗cd70 car由以下部分构成:对cd70特异的抗cd70单链可变片段(scfv),随后是与4-1bb的细胞内共信号传导结构域和cd3ζ信号传导结构域融合的cd8铰链和跨膜结构域。
[0267]
ctx130的示例性生产过程在图3a中示出。
[0268]
生产过程的演变
[0269]
基于由实例1至4中描述的优化过程确定的条件,ctx130生产过程以三种生产规模进行,包括研究规模、开发规模和临床规模。研究规模过程以小规模进行,并且研究规模过程按比例放大并转移到开发规模过程和临床规模过程。为了可行性和操作参数的调整,使用实验室级原材料对药物物质开展初始计划(4批)。随后,对临床规模过程实施gmp来源的原材料(sgrna、cas9和raav-145b)和定量验收标准的使用,该临床规模过程在操作上与开发规模过程相同。
[0270]
原材料的选择
[0271]
用于制备ctx130的原材料包括:
[0272]-从健康供体收集的白细胞单采样,
[0273]-细菌来源的cas9核酸酶,
[0274]-三种单引导rna(sgrna),即靶向cd70基因座的cd70-7 sgrna、靶向trac基因座的ta-1和靶向β2m基因座的β2m-1,以及
[0275]-编码抗cd70 car基因的重组aav-6载体(raav-145b)。
[0276]
用于进行ctx110的基因修饰的组分以及经过编辑的trac和β2m基因座的结构信息提供如下:
[0277]
cas9核酸酶的氨基酸序列(seq id no:1):
[0278][0279]
表8.sgrna序列和靶基因序列。
[0280][0281]
*指示具有2
′‑
o甲基硫代磷酸酯修饰的核苷酸。
[0282]“n”是指5
′
末端处的间隔子序列。
[0283]
表9.经过编辑的trac基因序列。
[0284][0285]
表10.经过编辑的β2m基因序列。
[0286][0287]
表11.经过编辑的cd70基因序列。
[0288][0289]
表12.抗cd70 car构建体组分的序列。
[0290]
[0291]
[0292]
[0293]
[0294]
[0295][0296]
ctx130的生产过程描述
[0297]
(i)t细胞富集
[0298]
使用自动化细胞处理系统,使用抗cd8和抗cd4抗体包被的磁珠的混合物,通过磁性分离从白细胞单采材料(白细胞单采样)中富集t细胞。富集前,对白细胞单采样进行取样以确定细胞计数和活力(≥80%)。
[0299]
在含有hsa的pbs/edta缓冲液中分离富集的细胞,然后对富集的细胞进行取样以确定细胞计数、活力(≥80%)、t细胞纯度(≥70%cd3)和无菌性。之后将细胞在4℃
±
1℃下离心并以50x106个活细胞/ml的目标浓度重悬于cryostor cs5中。
[0300]
(ii)t细胞冻存
[0301]
对细胞进行取样以确定细胞计数、活力(≥80%),然后以2,500x106个细胞/袋的靶细胞数将细胞等分到醋酸乙烯乙酯冻存袋中(30至70ml细胞悬液)。一次白细胞单采样可能足以产生1至2袋t细胞。将每个袋热封,贴标签,在2℃至8℃下储存直至转移至可控速率冷冻机,随后转移至气相液氮中储存。
[0302]
(iii)t细胞解冻、第一次电穿孔和激活
[0303]
将一袋冷冻的富集的t细胞解冻,转移到3l袋中并稀释到补充的x-vivo
tm 15培养基(x-vivo
tm 15,5%人血清、100iu/ml rhil2、100iu/ml rhil7)中。对细胞进行取样以确定细胞计数和活力(≥70%)。
[0304]
将细胞在20℃
±
1℃下以540g离心15分钟。将细胞沉淀物重悬于电穿孔缓冲液中并在相同条件下再次离心。将细胞再次重悬于电穿孔缓冲液中达到300x106个细胞/ml的目标浓度。
[0305]
将cas9核酸酶与cd70-7 sgrna在微量离心管中混合并在室温下温育不少于10分钟以形成核糖核蛋白(rnp)复合物。然后将cas9/sgrna与细胞混合,使cas9和cd70-7 sgrna分别达到0.15mg/ml和0.16mg/ml的最终浓度。
[0306]
将混合物等分并通过移液加载到电穿孔盒中。将盒加盖并使用基于流式电穿孔的转染系统依次对盒进行电穿孔。
[0307]
电穿孔后,将来自每个盒的细胞汇集在125ml锥形瓶中并在37℃下温育不少于20分钟。对细胞进行取样以确定活力(≥50%)和计数。然后以1:12.5(v/v)的比率添加与重组人源化cd3和cd28激动剂缀合的可溶性胶体聚合物纳米基质溶液以激活细胞。
[0308]
将细胞以目标密度2x106个活细胞/ml接种于静态细胞培养容器中,每个容器具有总体积为大约500ml的补充的x-vivo
tm 15培养基/与重组人源化cd3和cd28激动剂缀合的胶体聚合物纳米基质。
[0309]
将静态细胞培养容器在37℃
±
1℃和5%
±
1%co2下温育72
±
4小时。在整个过程中,每当处理静态细胞培养容器时,都检查它们的裂缝和泄漏,以及透明黄色培养基的存在。
[0310]
(iv)稀释
[0311]
三(3)天后,将补充的x-vivo
tm 15培养基添加到每个静态细胞培养容器中达到5l的最终体积。将细胞进一步在37℃
±
1℃和5%
±
1%co2下温育过夜。
[0312]
(v)第二次电穿孔和转导
[0313]
使用连接至静态细胞培养容器液浸管的泵将补充的x-vivo
tm 15培养基的体积减少至大约500ml的最终体积。
[0314]
轻轻涡旋静态细胞培养容器以使细胞重悬于培养基中。对细胞进行取样以确定细胞计数、活力(≥70%)。
[0315]
将细胞转移至500ml离心管中,并在20℃
±
1℃下以540g离心15分钟。将细胞沉淀物重悬于电穿孔缓冲液中并在相同条件下再次离心。将细胞再次重悬于电穿孔缓冲液中达到300x106个细胞/ml的目标浓度。
[0316]
将cas9核酸酶与ta-1sgrna和β2m-1sgrna在单独的微量离心管中混合。将每种溶液在室温下温育不少于10分钟以形成每种核糖核蛋白(rnp)复合物。将这两种cas9/sgrna混合物组合,并且与细胞混合,使cas9、ta-1和β2m-1分别达到0.3mg/ml、0.08mg/ml和0.2mg/ml的最终浓度。
[0317]
将混合物等分并通过移液加载到电穿孔盒中。将盒加盖并使用基于流式电穿孔的转染系统依次对盒进行电穿孔。
[0318]
电穿孔后,将来自每个盒的细胞汇集在125ml锥形瓶中并在37℃下温育不少于20分钟。对细胞进行取样以确定活力(≥70%)和计数。用x-vivo
tm 15培养基将细胞稀释至
1x107个细胞/ml的目标,并以20,000至50,000vg/细胞的moi添加新鲜解冻的raav-145b。将细胞在37℃、5%co2下温育不少于60分钟。
[0319]
(vi)细胞扩增
[0320]
将细胞用补充的x-vivo
tm 15培养基稀释,对细胞进行取样以确定细胞活力(≥70%)和计数,并将细胞以0.2x106个活细胞/cm2至0.5x106个活细胞/cm2之间的密度接种到两个静态细胞培养容器以及一个较小的静态细胞培养容器(充当卫星培养物以用于细胞监测)。在37℃
±
1℃和5%
±
1%co2下温育静态细胞培养容器。
[0321]
将细胞培养物温育长达9天。在此期间,每3至4天用每ml培养体积100iu的rhil2和rhil7补充培养物。
[0322]
在整个扩增过程中测试卫星细胞培养物的细胞计数、活力和t细胞纯度。当卫星培养物中的细胞密度达到大约30x106/cm2时,进行tcrαβ耗尽。如果卫星细胞密度未达到30x106/cm2,则在第9天对主培养物进行tcrαβ耗尽。
[0323]
(vii)tcrαβ耗尽
[0324]
使用连接至静态细胞培养容器液浸管的泵将每个静态细胞培养容器的培养基减少至大约500ml的最终体积。在去除大部分培养基后,轻轻涡旋静态细胞培养容器以将细胞重悬于培养基中。
[0325]
将细胞转移到装有与静态细胞培养容器连接的液浸管的500ml离心管中。对细胞进行取样以确定活力(≥70%)、计数和%car。然后将细胞在20℃
±
1℃下以540g离心15分钟。将细胞沉淀物重悬并汇集在少于650ml含有0.5%hsa的pbs/edta中。将细胞悬液转移到与自动化细胞处理系统连接的无菌袋。自动化细胞处理系统将细胞与生物素缀合的抗tcrαβ抗体一起温育。将细胞洗涤并与抗生物素磁珠一起温育以允许使用自动化细胞处理系统耗尽tcrαβ
+
细胞。
[0326]
测试细胞的细胞计数、活力(≥70%)和%car细胞。
[0327]
(viii)细胞恢复
[0328]
将耗尽的细胞重悬于补充的x-vivo
tm 15培养基中并转移到3l袋中,接种到静态细胞培养容器中并在37℃
±
1℃和5%
±
1%co2下温育过夜。
[0329]
(ix)细胞收获(药物物质)
[0330]
为了收获细胞,从培养箱中取出静态细胞培养容器,并让其静置以使细胞沉降。使用泵从每个静态细胞培养容器中去除生长培养基达到大约500ml的最终体积。对去除的培养基进行取样以确定无菌性。
[0331]
轻轻涡旋静态细胞培养容器以使细胞重悬于培养基中。使用泵将每个静态细胞培养容器的内容物转移到3l转移袋中,并取样进行浓度、活力和药物物质批次放行检测。然后将细胞通过40μm输血过滤器在重力作用下过滤到单独的无菌3l袋中。
[0332]
ctx130的表征
[0333]
ctx130是一种cd70定向的t细胞免疫疗法,其由表达抗cd70 car并且具有基因破坏的cd70、trac和β2m基因的同种异体t细胞构成。进行非临床药理学和毒理学研究以表征ctx130的非gmp开发批次的潜在功效和毒性。
[0334]
ctx130的非gmp开发批次的生产和表征
[0335]
本研究的目的是确定使用本文所述的方法是否实现非gmp cd70 car t细胞的可
再现生产。
[0336]
编辑三个单独的人t细胞供体以产生ctx130的非gmp开发批次,其中在初始步骤中使用含有cas9和针对cd70的grna的rnp,随后在第二步骤中使用含有cas9及针对trac和β2m的grna的rnp,接着用含有编码car的供体模板的aav6转导。随后使用柱纯化耗尽细胞中剩余的残余tcr
+
细胞。
[0337]
简言之,将来自3个单独供体的t细胞解冻并用含有cas9和靶向cd70基因座的grna的rnp对t细胞进行电穿孔,然后使用缀合至重组人源化cd3和cd28激动剂的胶体聚合物纳米基质激活t细胞3天。在第4天,将珠稀释并使t细胞再扩增一天。在第5天,用含有cas9及靶向trac和β2m基因座的grna的rnp对细胞进行电穿孔,随后将细胞与包含含有cd70car的hdr模板的aav6一起温育。在第二基因编辑步骤后十天,使用流式细胞仪分析细胞以评价trac、β2m和cd70的敲除效率以及表达car的细胞的百分比。使用抗trac、β2m和cd70蛋白的抗体进行染色,同时通过用生物素标记的抗小鼠fab2抗体染色,随后与荧光链霉亲和素一起温育来检测car表达。
[0338]
对经过编辑的细胞的分析显示出99.7%
±
0.1%trac阴性细胞、79.4%
±
1.1%β2m阴性细胞和98.9%
±
0.3%cd70-细胞(表13)。在3个测试供体的80.8%
±
8.4%细胞中检测到car表达(表13)。使用第四供体(供体4)利用相同的过程产生ctx130的另外研究批次,但是该研究批次没有耗尽剩余的残余tcr
+
细胞。
[0339]
表13.来自4个单独供体的ctx130批次的编辑效率总结。
[0340]
样品%tcr-%β2m-%cd70-%car
+
供体199.680.6399.1585.2供体299.878.8198.6271.1供体399.978.799.0686.1平均值99.7
±
0.179.4
±
1.198.9
±
0.380.8
±
8.4供体4*99.485.990.279
[0341]
*在没有耗尽残余tcr
+
细胞的情况下产生的ctx130研究批次;不包括在平均值中。
[0342]
(i)效应细胞因子释放
[0343]
本研究的目的是评估ctx130细胞在与cd70
+
或cd70-细胞共培养时分泌干扰素γ(ifnγ)和白介素2(il-2)的能力。
[0344]
将人靶细胞(cd70
+
细胞系a498和achn,以及cd70-细胞系mcf7)与t细胞以不同比率(从0.125:1至4:1的t细胞与靶细胞之比)按96孔板中每孔50,000个靶细胞共培养24小时。将靶细胞与ctx130细胞或对照细胞(未编辑的t细胞)一起温育。测量培养基上清液中ifnγ和il-2的水平并证实ctx130在与cd70
+
共培养时具有分泌ifnγ和il-2的能力,但在与cd70-细胞共培养时不具有分泌ifnγ和il-2的能力。
[0345]
(ii)肿瘤细胞细胞毒性
[0346]
本研究的目的是评估ctx130细胞杀伤cd70
+
细胞的能力。简言之,将人靶cd70
+
细胞(a498和achn)以每孔50,000个靶细胞铺板在96孔板中过夜,然后与ctx130或未编辑的t细胞以不同比率(从0.125:1至4:1的t细胞与靶细胞之比)共培养24小时。测量靶细胞的杀伤并证实ctx130细胞在体外杀伤cd70
+
细胞系。
[0347]
(iii)其他研究
[0348]
其他研究显示了ctx130细胞在肾细胞癌和塞扎里综合征的皮下模型中限制肿瘤细胞生长的能力,并证实了小鼠在包括存活、gvhd临床体征和体重的每个测量终点方面均对ctx130治疗耐受良好。
[0349]
(iv)人体组织交叉反应性
[0350]
本研究的目的是在基于免疫组织化学的组织交叉反应性研究中评价ctx130中所含的抗cd70 car的选择性。本研究中使用的供试品是ctx130的scfv部分来源的抗体。在以下两种抗体浓度下评价32种人体组织的标准组:最佳浓度(2.5μg/ml)和高浓度(10.0μg/ml),以试图捕获与人体组织的任何潜在结合。对于每种所测试的组织,评价来自3个供体的切片。在一些淋巴组织(淋巴结和扁桃体)中观察到极少至中度的阳性染色,这与正常cd70表达模式一致。在该组的剩余组织中未观察到染色。在阳性对照(人肾细胞癌肿瘤细胞)中观察到强染色。
[0351]
(v)细胞因子非依赖性生长
[0352]
本研究的目的是评估ctx130在不存在血清及细胞因子il-2和il-7的情况下增殖的能力。简言之,来自研究批次和非gmp开发批次的ctx130细胞在完全t细胞培养基、含有血清但不含il2或il7细胞因子(仅血清)的培养基或者不含血清或细胞因子的培养基(基础培养基)中生长。第0天发生在基因组编辑后14天。对于研究批次和非gmp开发批次,在不存在细胞因子的情况下没有观察到生长。这些结果表明在基因组编辑后无血清和细胞因子的培养基中缺乏生长和增殖。
[0353]
实例6:改进的细胞扩增
[0354]
用于增加ctx130细胞扩增产量的优化电穿孔
[0355]
本披露中所述的方法利用电穿孔将各种核酸和多肽递送至受体t细胞,包括例如包含cas9和引导rna复合物的各种核糖核蛋白(rnp)复合物。电穿孔过程中使用的仪器没有特别限制,因为来自不同制造商的任何合适的电穿孔仪器都可用于本文所述的方法中。电穿孔中使用的细胞接种密度没有特别限制。
[0356]
本实例使用能够对盒中增加数量的细胞进行电穿孔的电穿孔仪器,这些盒能够在保持有效编辑的同时保留较大体积。更大的电穿孔容量通过为转导和扩增提供更大数量的经过编辑的细胞来增加任何给定工程化t细胞(例如ctx130工程化t细胞产物)的产量,例如增加多达两倍。这在生产中是有利的,因为获得这种增加的容量不需要延长过程持续时间和/或细胞倍增。
[0357]
例如,另外的细胞可用于接种另外的t细胞培养容器(具有5000ml培养基容量的500cm2透气膜表面积),例如2个或更多个另外的培养容器。例如,随着细胞数目的增加,可接种最多4x培养容器,其中300e6≤x≤600e6细胞可接种在2x培养容器中,600e6≤x≤800e6细胞可接种在3x培养容器中,或≤800e6细胞可接种在4x培养容器中。
[0358]
在一些方面,每个培养容器接种约400,000个细胞/cm2与500,000个细胞/cm2之间。替代性地,每个培养容器接种约250,000个细胞/cm2与500,000个细胞/cm2之间,每个培养容器接种约300,000个细胞/cm2与500,000个细胞/cm2之间,每个培养容器接种约150,000个细胞/cm2与250,000个细胞/cm2之间,每个培养容器接种约150,000个细胞/cm2与500,000个细胞/cm2之间,每个培养容器接种约150,000个细胞/cm2与600,000个细胞/cm2之间。
[0359]
在一些方面,目标接种密度为至少约150,000个细胞/cm2或至少约250,000个细
胞/cm2或至少约300,000个细胞/cm2或至少约400,000个细胞/cm2或至少约500,000个细胞/cm2。
[0360]
在一些方面,目标接种密度为约250,000个细胞/cm2。在其他方面,目标接种密度为约500,000个细胞/cm2。
[0361]
可以使用能够保留高达1ml体积的电穿孔盒。使用该系统,可以在多达七个g1000盒中对2.7x109个细胞进行电穿孔。使用与3ml注射器连接的一次性钝头针头从盒中回收细胞也将消除微量移液管吸头弹射到锥形瓶中的风险。
[0362]
使用具有较大容量的系统也有利于细胞转导步骤。将用于转导的7e8个细胞的当前最大值加倍到1.4e9个细胞产生了足够的材料以接种多达四个用于扩增的细胞培养容器。因此,可以保持固定的第9天耗尽,从而在相同量的处理时间内有效地使每次运行的产量最多加倍。
[0363]
ctx130生产过程中的其他步骤如以上实例中所述。
[0364]
等同适用
[0365]
尽管本文已经描述并展示了几个发明实施例,但本领域普通技术人员将容易想象用于执行功能和/或获得结果和/或本文描述的优点中的一个或多个的多种其他装置和/或结构,并且此类变型和/或修改中的每一个被视为是在本文描述的发明实施例的范围内。更一般地说,本领域的技术人员将容易理解,本文描述的所有参数、尺寸、材料以及配置意味着为示例性的,并且实际参数、尺寸、材料和/或配置将取决于发明传授内容所用于的一种或多种具体应用。本领域的技术人员仅仅使用常规实验将认识到或能够确认本文描述的具体发明实施例的许多等同物。因此,应了解,前述实施例是仅借助于实例来呈现,并且在所附权利要求书和其相等形式的范围内,发明实施例可以按与具体地描述和要求不同的方式实践。本披露的发明实施例涉及本文描述的每个单独特征、系统、物品、材料、试剂盒和/或方法。此外,如果此类特征、系统、物品、材料、试剂盒和/或方法并不相互矛盾,两个或更多个此类特征、系统、物品、材料、试剂盒和/或方法的任意组合包括在本披露的发明范围内。
[0366]
如本文限定并使用的所有定义应当被理解成凌驾于所限定术语的字典定义、通过援引而并入的文件中的定义、和/或一般含义。
[0367]
本文披露的所有参考文献、专利和专利申请都相对于每个被引用的主题通过援引而并入,这在一些情况下可以包括文件的全部内容。
[0368]
除非清楚地作相反指示,否则在说明书中和在权利要求中,如本文使用的不定冠词“一个”和“一种”(a和an)应当理解为意思指“至少一个(种)”。
[0369]
在说明书中和在权利要求中,如本文使用的短语“和/或”应当理解为意思指如此联在一起的要素中的“任何一个或两个”,即,要素在一些情形中联合地存在并且在其他情形中分离性地存在。用“和/或”列出的多个元素应当以相同的方式来解释,即,这样结合的元素中的“一个或多个”。除了由“和/或”从句具体鉴定的元素,其他元素可以任选地存在,无论是与具体鉴定的那些元素相关还是不相关。因此,作为非限制性实例,当结合开放式语言诸如“包括”使用时,对“a和/或b”的提及可以在一个实施例中仅指a(任选地包括除了b的元素);在另一个实施例中,仅指b(任选地包括除了a的元素);在又另一个实施例中,指a和b(任选地包括其他元素);等。
[0370]
在说明书中和在权利要求中,如本文使用的“或”应理解为具有与如以上所定义的“和/或”相同的含义。例如,当分隔一个清单中的项目时,“或”或“和/或”应当解释为包容性的,即,包括多个元素或元素清单中的至少一个,而且包括其中的多于一个,以及任选地,另外的未列出的项目。仅相反地清楚指示的术语,诸如
“……
中的仅一个”或
“……
中的恰好一个”,或者当用于权利要求中时,“由
……
组成”将指恰好包括一些或一列元素中的一个元素。总体而言,如本文使用的术语“或”当前面加有排他性的术语,诸如“任何一个”、
“……
中一个”、
“……
中仅一个”或
“……
中只有一个”时,应当仅解释为指示排他性的替代形式(即,“一个或另一个,但非两者”)。“主要由
……
组成”当用于权利要求中时,应具有如在专利法领域中所用的其普通含义。
[0371]
如本文在说明书中和在权利要求中所用,关于一个或多个要素的列表的短语“至少一个”应理解为意指选自该要素列表中的任一个或多个要素的至少一个要素,但不一定包括在该要素列表内具体地列出的每个和每一个要素中的至少一个,并且不排除该要素列表中的多个要素的任何组合。这一定义还允许,可以任选地存在除该短语“至少一个”所指元素清单内具体地鉴别出的元素外的元素,无论是与具体地鉴别出的那些元素相关还是不相关。因此,作为非限制性实例,“a和b中的至少一个”(或等效地,“a或b中的至少一个”,或等效地“a和/或b中的至少一个”)在一个实施例中可以是指至少一个、任选地包括多于一个a,而不存在b(并且任选地包括除了b的元素);在另一个实施例中,可以是指至少一个、任选地包括多于一个b,而不存在a(并且任选地包括除了a的元素);在又另一个实施例中,可以是指至少一个、任选地包括多于一个a,以及至少一个、任选地包括多于一个b(并且任选地包括其他元素);等。
[0372]
还应当理解,除非明确指出相反,在包括多于一个步骤或动作的本文所要求保护的任何方法中,方法的步骤或动作的顺序不一定限于其中方法的步骤或动作被列举的顺序。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1