一种食品安全级枯草芽孢杆菌及其在生产几丁二糖脱乙酰酶中的应用
本发明涉及一种食品安全级枯草芽孢杆菌及其在生产几丁二糖脱乙酰酶中的应用,属于生物分子改造及基因工程技术领域。
背景技术:
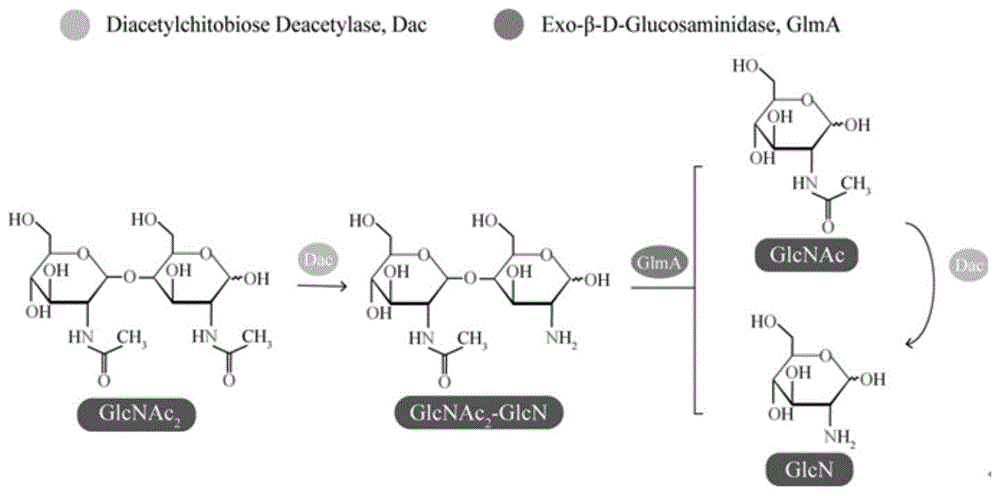
几丁二糖脱乙酰酶dac属于糖类酯酶,对乙酰氨基葡萄糖单体依然具有很高的催化活性,在代谢途径中首先作用的底物为几丁寡糖glcnacn或几丁二糖glcnac2,从glcnacn或glcnac2的还原端水解下乙酰基团,生成非还原性末端部分乙酰化的glcnac-glcn后,在外切氨基葡萄糖苷酶glma的水解作用下,将glcnac-glcn水解成n-乙酰氨基葡萄糖glcnac和氨基葡萄糖glcn,此后,几丁二糖脱乙酰酶dac再作用于生成的n-乙酰氨基葡萄糖glcnac单体,将其水解成氨基葡萄糖glcn。
相较于目前市场上存在的多针对以寡聚体或多聚体形式存在的乙酰氨基葡萄糖有催化活性的脱乙酰酶,来源于极端嗜热古菌pyrococcushorikoshii的几丁二糖脱乙酰酶dac可以利用n-乙酰氨基葡萄糖glcnac为底物进行单酶催化反应,实现氨基葡萄糖glcn的一步高效生产。
氨基葡萄糖glcn是一种重要的功能性单糖,在生物体的生长过程中起着不可替代的作用。作为组成黏膜分泌物、结缔组织、皮肤、肌腱、韧带和软骨等必不可少的成分,氨基葡萄糖glcn在许多国家已被列为医药品行列。氨基葡萄糖glcn除了在治疗软骨性关节炎方面有着良好的作用外,对肝癌细胞的生长也有一定的抑制作用。
目前,氨基葡萄糖glcn在许多国家已被列入医药品行列,其生产方式一般为甲壳素水解法和微生物发酵法。其中,甲壳素水解法常用强酸热解以脱除乙酰基,容易产生大量的环境污染物,而且原料会受到地域和季节的限制,获得的产品中乙酰基的脱除程度不一致,导致产品均一性差。微生物发酵法虽然克服了化学水解法带来的严重污染,但发酵液中难以实现氨基葡萄糖glcn的高浓度积累。由于氨基葡萄糖glcn存在广泛的市场,因此,探寻一种更高效、更安全的氨基葡萄糖glcn生产方法势在必行。
枯草芽胞杆菌作为一种典型的模式工业微生物,具有遗传背景清晰、基因编辑成熟等优点。随着对枯草芽孢杆菌遗传调控机制的不断揭示,作为异源蛋白表达“细胞工厂”的枯草芽胞杆菌已被证实基因组上包含很多对生长和异源酶表达不利的非必需基因,如:前噬菌体、菌体自溶、芽胞形成、异源酶转录负调控因子等基因,现如今通过对其基因组进行删减的手段,合理设计简化细胞内的调控代谢网络,为细胞减负的同时提高宿主对异源酶的生产效率,使其成为理想的底盘细胞。
例如记载于公开号为cn109777761b的中国发明专利中,公开了一种分泌表达几丁二糖脱乙酰酶dac的工程菌及其构建方法,通过融合表达几丁二糖脱乙酰酶dac与yncm信号肽,并获得了5’端的非翻译区突变体,发酵60h胞外酶活最高为1548.7u/ml。
但是,目前的几丁二糖脱乙酰酶dac的酶活较低,并且现有的几丁二糖脱乙酰酶dac工程菌仍难以满足工业化利用酶法制备氨基葡萄糖glcn的最低酶活需求,氨基葡萄糖glcn在转化过程中的产量和转化率还较低,所以需要进一步提高氨基葡萄糖glcn的产量。
技术实现要素:
为解决现有技术中存在的几丁二糖脱乙酰酶dac产量低、活性低、工程菌株传代不稳定、生长受限等问题,本发明旨在通过信号肽的筛选及优化,提高几丁二糖脱乙酰酶dac的分泌效率;针对目标蛋白在枯草芽孢杆菌表达系统中的优化改良,多是从转录、翻译、折叠、转运以及菌株改造等方面进行模块化的改造及组装,其中蛋白的转运与信号肽这一分泌元件有着密不可分的关系,由于对分泌机制了解有限,准确预算出一个蛋白的最佳信号肽还较为困难,现如今对信号肽的应用仍然处于探索阶段。虽然研究人员开发了有助于信号肽选择的预测工具,但预测数值和蛋白的实际分泌量似乎不成线性关系。
本发明通过信号肽的筛选及优化,底盘细胞的优化:以bacillussubtilis168为出发菌株,引入crispr/cpf1无痕编辑系统对底盘细胞npre、apre、nprb、bpr、mpr、epr6种胞外蛋白酶进行无义突变,同时将底盘细胞生长的非必需基因coma、oppa基因进行敲除,通过提高生物量来进一步提高氨基葡萄糖glcn的产量,提升几丁二糖脱乙酰酶酶活。
本发明提供了一种重组枯草芽孢杆菌,所述重组枯草芽孢杆菌以p43nmk为载体,融合表达了几丁二糖脱乙酰酶与nprb信号肽,并在几丁二糖脱乙酰酶基因的5’非翻译区引入seqidno.3所示的dna片段,引入位点位于启动子p43的+1转录起始位点后的8个碱基之后;将核苷酸序列如seqidno.4所示的c4基因插入重组质粒中nprb信号肽的5’端。
在本发明的一种实施方式中,所述重组枯草芽孢杆菌以pp43nmkmut-c4-nprb-dac为载体,所述载体是在pp43nmkmut-c4-yncm-dac基础上,采用nprb信号肽替换yncm信号肽,所述pp43nmkmut-c4-yncm-dac载体公开于“几丁二糖脱乙酰酶的克隆表达与生物转化合成氨基葡萄糖的研究,姜竹,江南大学”论文当中。
在本发明的一种实施方式中,所述nprb信号肽的氨基酸序列如seqidno.1所示,其核苷酸序列如seqidno.6所示。
在本发明的一种实施方式中,所述nprb信号肽的氨基酸序列如seqidno.2所示(即nprb信号肽优化后的nprb5信号肽),其核苷酸序列如seqidno.7所示。
在本发明的一种实施方式中,几丁二糖脱乙酰酶来源于极端嗜热古菌,编码所述几丁二糖脱乙酰酶的基因序列如seqidno.5所示。
在本发明的一种实施方式中,所述p43nmk载体上的rbs序列gtaagagaggaatgtacac突变为ggaagggaggaatagagac。
在本发明的一种实施方式中,所述重组枯草芽孢杆菌以bacillussubtiliswb600为宿主。
在本发明的一种实施方式中,所述重组枯草芽孢杆菌的宿主细胞为:以bacillussubtilis168为出发菌株,敲除bacillussubtilis168基因组上的npre、apre、nprb、bpr、mpr、epr6个胞外蛋白酶及coma和oppa基因。
在本发明的一种实施方式中,所述npre、apre、nprb、bpr、mpr、epr基因在ncbi的登录号分别为:935981、939313、936384、939695、938430、937332;所述coma和oppa基因在ncbi的登录号分别为:937179、936398。
本发明还提供了一种制备几丁二糖脱乙酰酶的方法,所述方法应用上述重组枯草芽孢杆菌进行发酵制备。
在本发明的一种实施方式中,以2~6%(v/v)的接种量将重组枯草芽孢杆菌的种子液接种到tb发酵培养基中,培养条件为:30-40℃、160-260rpm培养45-85h。
本发明还提供了构建上述重组枯草芽孢杆菌的方法,所述方法包括以下步骤:
(1)制备重组载体,所述重组载体是在pp43nmkmut-c4-yncm-dac基础上,采用nprb信号肽替换yncm信号肽;得到重组表达载体pp43nmkmut-c4-nprb-dac;所述nprb信号肽的氨基酸序列如seqidno.1所示,或所述nprb信号肽的氨基酸序列如seqidno.2所示
(2)将步骤(1)制备得到的重组载体转入枯草芽孢杆菌,得到重组枯草芽孢杆菌。
在本发明的一种实施方式中,所述重组载体pp43nmkmut-c4-nprb-dac上的rbs序列gtaagagaggaatgtacac突变为ggaagggaggaatagagac。
在本发明的一种实施方式中,所述重组枯草芽孢杆菌以枯草芽孢杆菌wb600为宿主。
在本发明的一种实施方式中,所述重组枯草芽孢杆菌的宿主细胞为:以bacillussubtilis168为出发菌株,敲除npre、apre、nprb、bpr、mpr、epr6个胞外蛋白酶及coma和oppa基因。
本发明还提供了一种制备氨基葡萄糖的方法,所述方法采用上述重组枯草芽孢杆菌,或上述方法制备得到几丁二糖脱乙酰酶为催化剂,以n-乙酰氨基葡糖为底物,制备得到氨基葡萄糖。
在本发明的一种实施方式中,所述底物终浓度为65~185g/l。
在本发明的一种实施方式中,所述方法的反应条件为:在30-60℃条件下反应2-62min。
在本发明的一种实施方式中,所述方法为,采用上述重组枯草芽孢杆菌的发酵上清液为催化剂,所述发酵上清液,是将种子培养液按照2~6%(v/v)的接种量接到发酵培养基中,37~40℃培养45-85h,经10000-16000rpm、4℃离心1-5min。
在本发明的一种实施方式中,所述种子培养液为,在30-40℃条件下培养至od600为3.0-6.0的重组枯草芽孢杆菌。
在本发明的一种实施方式中,所述发酵培养基成分:蛋白胨12g/l、酵母提取物24g/l、甘油4ml,kh2po42.31g/l和k2hpo412.54g/l。
本发明还提供了上述重组枯草芽孢杆菌在制备药物或膳食补充剂中的应用。
本发明还提供了上述重组枯草芽孢杆菌在制备氨基葡萄糖或含氨基葡萄糖的产品中的应用。
本发明还提供了一种提高几丁二糖脱乙酰酶dac分泌效率的方法。
本发明还提供了一种提高几丁二糖脱乙酰酶dac表达量的方法。
本发明还提供了一种提高几丁二糖脱乙酰酶dac工程菌株生长速率的方法。
本发明还提供了一种bacillussubtiliszr6aa/pp43nmkmut-c4-nprb5-r2-dac工程菌株的构建方法及其应用。
有益效果
(1)本发明通过对几丁二糖脱乙酰酶dac的表达载体pp43nmkmut-c4-yncm-dac的信号肽进行筛选和优化,并且对底盘细胞进行了优化,提高几丁二糖脱乙酰酶dac的分泌效率,提高酶活,构建了可以稳定高效表达几丁二糖脱乙酰酶dac的食品安全级工程菌株。与现有技术相比,几丁二糖脱乙酰酶胞外酶活得到了极大的提高。本转化方法简单易行,条件温和,高效环保,产物得率高。此法有利于解决传统合成法中污染严重,产率低下等问题,而且此法易于控制,利于推广应用。
(2)本发明在bacillussubtiliswb600/pp43nmkmut-c4-yncm-dac重组菌株的基础上进一步改造,替换原始信号肽,构建的bacillussubtiliswb600/p43nmkmut-c4-nprb-dac重组菌株发酵60h胞外酶活最高达3168u/ml,通过对信号肽优化,构建的bacillussubtiliswb600/p43nmkmut-c4-nprb5-dac重组菌株发酵60h胞外酶活最高达3682.2u/ml。
(3)本发明在bacillussubtiliswb600/p43nmkmut-c4-nprb5-dac重组菌株的基础上进一步改造,通过将所述重组载体pp43nmkmut-c4-nprb5-dac上的rbs序列gtaagagaggaatgtacac突变为ggaagggaggaatagagac,获得bacillussubtiliswb600/p43nmkmut-c4-nprb5-r2-dac重组菌株,该菌株发酵60h胞外酶活达4012.3u/ml。
(4)本发明通过对宿主细胞进行改造,以bacillussubtilis168为出发菌株,对底盘细胞npre、apre、nprb、bpr、mpr、epr6种胞外蛋白酶进行无义突变,敲除影响菌株生长的coma、oppa基因,提高生物量,减缓发酵后期菌株的裂解死亡,构建得到重组菌株bacillussubtiliszr6aa/p43nmkmut-c4-nprb5-r2-dac,该菌株发酵60h胞外酶活最高达4774.2u/ml,较原始菌株提升了2.08倍。
附图说明
图1:本发明中几丁二糖脱乙酰酶dac催化反应过程。
图2:本发明中不同信号肽菌株构建示意图。
图3:本发明实施例1中不同信号肽引导几丁二糖脱乙酰酶dac分泌到胞外,胞外酶活及氨糖产量的比较。
图4:本发明实施例2中信号肽nprb的功能区域划分。
图5:本发明实施例2中信号肽nprb不同突变体引导几丁二糖脱乙酰酶dac分泌到胞外,胞外酶活及氨糖产量的比较。
图6:本发明实施例3中不同翻译强度的rbs序列对几丁二糖脱乙酰酶dac胞外酶活及氨糖产量的影响。
图7:本发明实施例3中所构建bacillussubtiliswb600/p43nmkmut-c4-nprb5-r2-dac与不产几丁二糖脱乙酰酶dac的bacillussubtiliswb600/p43nmk及原始生产几丁二糖脱乙酰酶dac的bacillussubtiliswb600/p43nmkmut-c4-yncm-dac的发酵上清液的sds-page分析情况;其中,泳道1,重组bacillussubtiliswb600/p43nmkmut-c4-nprb5-r2-dac的发酵上清液;泳道2,bacillussubtiliswb600/p43nmkmut-c4-yncm-dac的发酵上清液;泳道3:bacillussubtiliswb600/p43nmk的发酵上清液。
图8:本发明实施例5中以构建的新型底盘细胞bacillussubtiliszr6aa为表达宿主时,与以bacillussubtiliswb600为表达宿主时,几丁二糖脱乙酰酶dac胞外酶活的比较。
图9:本发明实施例5中以构建的新型底盘细胞bacillussubtiliszr6aa为表达宿主时,与以bacillussubtiliswb600为表达宿主时,细胞的生长情况比较。
具体实施方式
下面结合实施例和附图对本发明进行详细的解释说明。
下述实施例中所涉及的载体pp43nmkmut-c4-yncm-dac公开于“几丁二糖脱乙酰酶的克隆表达与生物转化合成氨基葡萄糖的研究,姜竹,江南大学”论文当中。
下述实施例中所涉及的检测方法:
几丁二糖脱乙酰酶酶酶活力检测方法:
通过邻苯二甲醛显色法进行粗酶液酶活检测,具体方式如下:
取发酵60h的发酵液1ml,以1200rpm,4℃,2min条件离心,上清液即为粗酶液,底物为100g/l的n-乙酰氨基葡萄糖glcnac溶液。将待测粗酶液与底物各100μl分装于1.5mlep管中,40℃预热5min。将预热后的100μl粗酶液加入100μl底物中,40℃震荡反应2min,精确计时,加入0.5mol/lhcl终止剂终止反应。以1200rpm,2min条件离心,取上清50μl于950μlpb1中,稀释20倍,混合均匀后取5μl加入100μl1×opa检测试剂中,震荡2min。利用酶标仪检测其在330nm处的吸光值。以失活后的酶液为反应空白对照。
氨基葡萄糖盐酸盐的标准曲线绘制,精确称取标准品(精确到0.0001g)配制0.5g/l、1g/l、2g/l、3g/l、4g/l、5g/l氨基葡萄糖盐酸盐溶液,取5μl加入100μlopa检测试剂中,震荡1min,30℃保温2min。利用酶标仪检测其在330nm处的吸光值,绘制标准曲线。
酶活力计算公式如下所示:
式中:x:粗酶液酶活力(u/ml);a:粗酶液体积0.1(ml);b:底物溶液体积0.1(ml);m:加入终止剂后样品稀释倍数1.5;c:反应液glcn浓度(g/l);d:空白液glcn浓度(g/l)。t:反应时间2(min);60:1h为60min;215.6:标准样品氨基葡萄糖盐酸盐摩尔质量(g/mol);0.1:底物溶液体积数(ml)x:酶液稀释倍数20。
所涉及的检测试剂如下:
底物n-乙酰氨基葡萄糖溶液(100g/l):5gn-乙酰氨基葡萄糖glcnac溶于pb1中,用pb1定容至50ml。
pb1磷酸钠缓冲液(200mmol/l,ph8.0):配制浓度为200mmol/l的nah2po4溶液和na2hpo4溶液。18mlnah2po4溶液与390mlna2hpo4溶液混匀。用ph测定仪测定ph值,确定ph8.0,室温储存。
pb2碳酸钠缓冲液(100mmol/l,ph10.5):配制浓度为100mmol/l的naco3溶液和nahco3溶液。18mlnah2po4溶液与390mlna2hpo4溶液混匀。用ph测定仪测定ph值,确定ph10.5,室温储存。
dtt二硫苏糖醇(2mol/l):称取308.5mg二硫苏糖醇于1mlddh2o中,混合均匀,密封低温保存。
10×opa检测试剂:500mg邻苯二甲醛opa加入100mlpb2中,混合均匀,密封低温避光保存。
1×opa检测试剂:3ml10×opa,300μl无水乙醇,15μldtt,加pb2定容到30ml,混匀后,密封低温避光保存。现用现配。
hcl终止剂(0.5mol/l):取1800μl盐酸于40mlddh2o中。
几丁二糖脱乙酰酶酶活力定义:在40℃反应条件下,1h能转化1μmol底物n-乙酰氨基葡萄糖glcnac为1μmol产物氨基葡萄糖glcn所需的酶量,称为一个酶活力单位,即1u=1μmol/h。
下述实施例中所涉及的培养基如下:
lb液体培养基(g/l):酵母粉5,蛋白胨10,nacl10,121℃灭菌15min。
lb固体培养基(g/l):酵母粉5,蛋白胨10,nacl10,琼脂粉20,121℃灭菌15min。
tb发酵培养基(g/l):酵母粉24,蛋白胨12,甘油4,kh2po42.31,k2hpo412.54,115℃灭菌20min。
实施例1:重组枯草芽孢杆菌的构建、几丁二糖脱乙酰酶dac的表达及信号肽的筛选具体步骤如下:
(1)选择同分泌途径的12条枯草芽孢杆菌内源信号肽,具体信号肽及其序列如表1所示:
表1:不同的信号肽及序列
(2)通过simplecloning法,如图2所示,以重组质粒pp43nmkmut-c4-yncm-dac(具体构建方法公开于“几丁二糖脱乙酰酶的克隆表达与生物转化合成氨基葡萄糖的研究,姜竹,江南大学”论文中)为模板,引物序列如表2所示,通过pcr扩增,经核酸电泳胶验证后转化大肠杆菌escherichiacolidh5α并送测,测序结果正确的转化子提质粒后,转入表达宿主枯草芽孢杆菌bacillussubtiliswb600。
表2:不同信号肽的引物序列
(3)使用接种环挑取携带不同信号肽的转化子在50ml离心管中进行种子培养。每管含有5ml添加卡那抗生素(10mg/ml)的lb液体培养基,抗生素添加体积为液体培养基体积的1‰,于37℃,220rpm,培养12h得到种子液;在500ml锥形瓶中,将制备得到的种子液以4%(v/v)的接种量转接至96ml添加卡那抗生素(10mg/ml)的tb液体培养基进行发酵培养,37℃、220rpm条件下培养60h,制备得到发酵液,将发酵液经4℃、12000rpm离心2min得到发酵上清液,即为粗酶液。
(4)对步骤(3)制备得到的粗酶液进行酶活的检测,结果如表3和图3所示。
(5)采用步骤(3)制备得到的发酵上清液为催化剂,取1ml发酵上清液,以终浓度为100g/l的n-乙酰氨基葡糖为底物,在40℃条件下反应2min,制备得到氨基葡萄糖盐酸盐,检测氨基葡萄糖盐酸盐的含量如表3和图3所示,其中control代表信号肽为yncm的实验结果(具体催化反应原理如图1所示)。
表3:含有不同信号肽的重组菌发酵制备得到几丁二糖脱乙酰酶的酶活及采用含有不同信号肽的重组菌株制备氨基葡萄糖(简称氨糖)的产量
通过锥形瓶发酵验证,信号肽nprb为本次筛选所得到的最优信号肽,如图3,在发酵60h后上清液的酶活峰值达3168u/ml,氨糖的产量高达158.4mol/l,为所有重组菌株的最高水平。
实施例2:几丁二糖脱乙酰酶dac优势信号肽的优化
对信号肽nprb进行优化,将氨基酸序列如seqidno.1所示的信号肽第1位的甲硫氨酸突变为赖氨酸,得到突变体m1k,命名为nprb1;
将氨基酸序列如seqidno.1所示的信号肽第1位的甲硫氨酸突变为赖氨酸,第3位的天冬酰胺突变为赖氨酸,得到突变体m1k-n3k,命名为nprb2;
将氨基酸序列如seqidno.1所示的信号肽第1位的甲硫氨酸突变为赖氨酸,第3位的天冬酰胺突变为赖氨酸,第5位的苏氨酸突变为赖氨酸,得到突变体m1k-n3k-t5k,命名为nprb3;
将氨基酸序列如seqidno.1所示的信号肽第1位的甲硫氨酸突变为赖氨酸,第3位的天冬酰胺突变为赖氨酸,第5位的苏氨酸突变为赖氨酸,第9位的亮氨酸突变为谷氨酰胺,得到突变体m1k-n3k-t5k-l9q,命名为nprb4;
将氨基酸序列如seqidno.1所示的信号肽第1位的甲硫氨酸突变为赖氨酸,第3位的天冬酰胺突变为赖氨酸,第5位的苏氨酸突变为赖氨酸,第9位的亮氨酸突变为谷氨酰胺,第11位的亮氨酸突变为天冬氨酸,得到突变体m1k-n3k-t5k-l9q-l11d,命名为nprb5;
涉及的引物序列如表4所示:
表4:不同突变体的引物序列
以pp43nmkmut-c4-nprb-dac为模板,采用上述引物序列,通过pcr扩增,分别构建得到含有5种突变体nprb1、nprb2、nprb3、nprb4、nprb5的重组载体pp43nmkmut-c4-nprb1-dac,pp43nmkmut-c4-nprb2-dac,pp43nmkmut-c4-nprb3-dac,pp43nmkmut-c4-nprb4-dac,pp43nmkmut-c4-nprb5-dac。
分别将上述重组载体按照实施例1的方法,以枯草芽孢杆菌bacillussubtiliswb600为表达宿主,构建重组菌,并进行发酵培养并进行酶活检测及检测制备得到的氨基葡萄糖的含量。
通过锥形瓶发酵验证,如图5和表5所示,其中control代表含有突变前的信号肽nprb的重组菌的实验数据。
表5:含有不同信号肽突变体的重组菌发酵制备得到几丁二糖脱乙酰酶的酶活及采用含有不同信号肽突变体的重组菌株制备氨基葡萄糖(简称氨糖)的产量
结果可以看出,含有信号肽突变体m1k-n3k-t5k-l9q-l11d,即nprb5的重组菌株在发酵60h后上清液的酶活峰值达3682.2u/ml,氨糖的产量高达184.11mol/l。
实施例3:rbs序列优化
对重组载体上的rbs进行优化,具体步骤如下:
以实施例2制备得到的pp43nmkmut-c4-nprb5-r0-dac(为了描述方便,将未优化之前的重组载体pp43nmkmut-c4-nprb5-dac,记为pp43nmkmut-c4-nprb5-r0-dac)为模板,引物如表6,进行pcr扩增,分别得到重组载体pp43nmkmut-c4-nprb5-r1-dac,pp43nmkmut-c4-nprb5-r2-dac,pp43nmkmut-c4-nprb5-r3-dac,pp43nmkmut-c4-nprb5-r4-dac,pp43nmkmut-c4-nprb5-r5-dac。
表6引物序列
分别将上述重组载体按照实施例1的方法,以枯草芽孢杆菌bacillussubtiliswb600为表达宿主,构建重组菌,并进行发酵培养并进行酶活检测,结果如图6和表7所示,其中control代表含有原始rbs序列的重组菌的实验数据。
表7:含有不同rbs序列的重组菌发酵制备得到几丁二糖脱乙酰酶的酶活及采用含有不同rbs序列的重组菌株制备氨基葡萄糖(简称氨糖)的产量
如图6所示,重组菌株bacillussubtiliswb600/p43nmkmut-c4-nprb5-r2-dac发酵60h胞外酶活最高,达4012.3u/ml,氨糖的产量高达200.615mol/l。
所获菌株bacillussubtiliswb600/p43nmkmut-c4-nprb5-r2-dac与原始生产几丁二糖脱乙酰酶dac的bacillussubtiliswb600/p43nmkmut-c4-yncm-dac及不产几丁二糖脱乙酰酶dac的bacillussubtiliswb600/p43nmk的sds-page分析情况如图7所示,其中1、2、3三条泳道分别对应bacillussubtiliswb600/p43nmkmut-c4-nprb5-r2-dac、bacillussubtiliswb600/p43nmkmut-c4-yncm-dac及bacillussubtiliswb600/p43nmk,从图7可以看到在31.8kda处有明显目的蛋白条带,证明几丁二糖脱乙酰酶dac得到了表达。
实施例4:枯草芽孢杆菌底盘细胞bacillussubtiliszr6aa的构建
具体步骤如下:
通过cas蛋白cpf1的dna内切酶活性实现crispr编辑,在引导rna(crrna)的介导下cpf1蛋白会在基因组的特定位点进行切割,造成双链断裂。
1、crrna载体构建
借助crrna在线设计网站crispr-dt设计特异性高的crrna,以减少脱靶现象的发生。以表8所列引物扩增待敲除基因coma、oppa的crrna片段,然后与载体pcrf17nm(载体pcrf17nm的构建方法公开于camers-b:crispr/cpf1assistedmultiple-geneseditingandregulationsystemforbacillussubtilis,yaokangwu,jiangnanuniversity论文中)分别进行goldengate组装,转化至escherichiacolidh5α,测序验证后表明成功构建4种crrna载体。
表8:引物序列
2、同源修复模板载体构建
扩增待敲除基因coma和oppa的上下游同源臂(序列如表9所示),借助碧云天一步克隆连接酶将上下游同源臂与步骤1中构建的对应crrna载体相连,构建敲除基因coma和oppa的同源修复模板载体,转化至escherichiacolidh5α,测序验证。
表9:coma和oppa的上下游同源臂
3、cas蛋白cpf1感受态细胞制备
质粒pht-xcr6(所述质粒pht-xcr6的构建方法公开camers-b:crispr/cpf1assistedmultiple-geneseditingandregulationsystemforbacillussubtilis,yaokangwu,jiangnanuniversity论文中)为cas蛋白cpf1表达载体,其中cpf1受木糖的诱导调控。
将质粒pht-xcr6转化至bszr600(bszr600的构建方法公开于camers-b:crispr/cpf1assistedmultiple-geneseditingandregulationsystemforbacillussubtilis,yaokangwu,jiangnanuniversity论文中,是一株以枯草芽孢杆菌168为出发菌株,敲除npre、apre、nprb、bpr、mpr、epr6个胞外蛋白酶,构建的一株6种胞外蛋白酶失活的枯草芽孢杆菌底盘细胞),然后再将阳性转化子制成感受态,具体步骤为:接种环挑取单菌落于2ml含有氯霉素(5mg/ml)抗性的液体lb培养基中,抗生素的添加体积为液体培养基体积的1‰,37℃,220rpm培养12h;测量od600,以液体培养基lb和木糖稀释至od600=1.0,木糖终浓度控制在3%,37℃,220rpm培养2h;此时菌株呈感受态状态,添加终浓度为10%的甘油后存于-80℃备用。
4、同源修复模板质粒转化
将步骤2中同源修复模板载体加入到步骤3中制备得到的cas蛋白cpf1感受态细胞中,37℃,220rpm培养2h;将菌液离心重悬于500μl含有氯霉素、卡那霉素和3%木糖的lb液体培养基中,37℃,220rpm培养12h,然后离心浓缩到150μl涂布添加有氯霉素、卡那霉素和3%木糖的lb固体培养基中,待lb固体培养基长出单菌落后进行菌落pcr验证是否完成基因编辑。
5、质粒的消除
以接种环挑取步骤4中制备得到的阳性菌落到添加有0.005%-0.006%十二烷基硫酸钠的无抗lb液体培养基中,37℃,220rpm培养6h后,进行无抗lb固体培养基划线,置于37℃培养箱培养12h后,挑取单菌落于含有氯霉素和卡那霉素的lb固体培养基上,进行质粒消除验证。质粒成功消除后获得新的枯草芽孢杆菌底盘细胞bacillussubtiliszr6aa;所述bacillussubtiliszr6aa菌株是以枯草芽孢杆菌168为出发菌株,敲除npre、apre、nprb、bpr、mpr、epr6个胞外蛋白酶,并且敲除coma和oppa基因。
实施例5:底盘细胞改造菌株发酵验证
具体步骤如下:
1、底盘细胞改造菌株感受态的制备
将实施例4中获得的新枯草芽孢杆菌底盘细胞bacillussubtiliszr6aa,参照实施例4的步骤3中cas蛋白cpf1感受态细胞制备方法,制备成感受态。
2、底盘细胞改造菌株发酵验证
将实施例3中制备得到的重组载体p43nmkmut-c4-nprb5-r2-dac,转化至bacillussubtiliszr6aa,获得bacillussubtiliszr6aa/p43nmkmut-c4-nprb5-r2-dac重组菌株;
按照实施例1的方法,将制备得到的重组菌株进行发酵并进行酶活检测,在发酵60h时,胞外酶活最高达4774.2u/ml,较原始菌株bacillussubtiliswb600/pp43nmkmut-c4-yncm-dac(图8中的空白对照1,其酶活为1548.7u/ml)提升了2.08倍,较实施例3中所获菌株bacillussubtiliswb600/p43nmkmut-c4-nprb5-r2-dac(图8中的空白对照2,其酶活为4012.3u/ml)提升了18.98%,结果如图8或表10所示,其中control-1表示:bacillussubtiliswb600/pp43nmkmut-c4-yncm-dac重组菌;control-2表示:bacillussubtiliswb600/p43nmkmut-c4-nprb5-r2-dac重组菌;△coma△oppa表示:bacillussubtiliszr6aa/p43nmkmut-c4-nprb5-r2-dac重组菌。
表10:不同重组菌发酵制备得到几丁二糖脱乙酰酶的酶活及其制备氨基葡萄糖(简称氨糖)的产量
重构底盘细胞后,重组菌株生长情况也得以改善,结果如图9所示,其中图9中的control代表bacillussubtiliswb600/p43nmkmut-c4-nprb5-r2-dac重组菌;△coma△oppa表示:bacillussubtiliszr6aa/p43nmkmut-c4-nprb5-r2-dac重组菌。
虽然本发明已以较佳实施例公开如上,但其并非用以限定本发明,任何熟悉此技术的人,在不脱离本发明的精神和范围内,都可做各种的改动与修饰,因此本发明的保护范围应该以权利要求书所界定的为准。
sequencelisting
<110>江南大学
<120>一种食品安全级枯草芽孢杆菌及其在生产几丁二糖脱乙酰酶中的应用
<130>baa210594a
<160>7
<170>patentinversion3.3
<210>1
<211>28
<212>prt
<213>人工序列
<400>1
metargasnleuthrlysthrserleuleuleualaglyleucysthr
151015
alaalaglnmetvalphevalthrhisalaserala
2025
<210>2
<211>28
<212>prt
<213>人工序列
<400>2
lysarglysleulyslysthrserglnleuaspalaglyleucysthr
151015
alaalaglnmetvalphevalthrhisalaserala
2025
<210>3
<211>73
<212>dna
<213>人工序列
<400>3
ggtaccattataggtaagagaggaatgtacacatggtcgtcaacatgttcgaggacatcg60
acacgttcgagga73
<210>4
<211>45
<212>dna
<213>人工序列
<400>4
atgaaaaaaatcacaacaaacgaacaatttaatgaactgattcaa45
<210>5
<211>819
<212>dna
<213>人工序列
<400>5
atggtcgtcaacatgttcgaggacatcgacacgttcgaggaagcgtttaacaagctgctg60
cgcgaagtcctggaatttgatctgcaaaatccgttcaaagacgcgaagaaagtcctttgc120
atcgaaccgcatccggacgattgcgttattggaatgggcggcacaatcaaaaaactgagc180
gatatgggcgtcgaagtcatctacgtttgcatgacagacggctatatgggcacaacagac240
gaaagcctgtcaggacacgaattagcagcaatccgccgcaaagaagaagaagaaagcgca300
cgcctgctgggcgttaaaaagatctattggctgaactaccgcgatacagaactgccgtat360
tcacgcgaagtccgcaaagatctgacgaaaattctgcgcaaagaacaaccggacggagtt420
tttgcaccagatccttggcttccgtacgaatcacatccggatcatagacgcacaggcttt480
ctggcgattgaatcagttgcgtttagccagctgccgaattttagcaacacggatctggac540
attggcctgaatccgtataacagcggaagctttatcgcgctgtactacacgcacaaaccg600
aactacatcgtcgacatcacggacctgatggaactgaaactgaaggcgattcgcgtccat660
agaagccagtttccggacgatatttgggagaaatgggaaccgttcctgagaacaatcgcg720
atgttctacggcgaaaaaatcggcgttcgctacggagaaggctttagaattatgccgggc780
ctgttctaccacatcacaccgtttacggacctgatctga819
<210>6
<211>84
<212>dna
<213>人工序列
<400>6
ttgcgcaacttgaccaagacatctctattactggccggcttatgcacagcggcccaaatg60
gtttttgtaacacatgcctcagct84
<210>7
<211>84
<212>dna
<213>人工序列
<400>7
aaacgcaaattgaaaaagacatctcagttagacgccggcttatgcacagcggcccaaatg60
gtttttgtaacacatgcctcagct84
- 还没有人留言评论。精彩留言会获得点赞!