制备(1R,3R)-2,2-二甲基-3-(Z)-丙-1-烯-1-基)环丙烷羧酸和酯的简短而有效的方法与流程
制备(1r,3r)-2,2-二甲基-3-(z)-丙-1-烯-1-基)环丙烷羧酸和酯的简短而有效的方法
技术领域
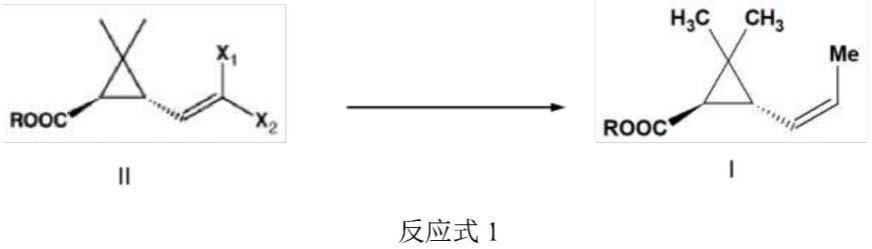
1.本发明涉及一种如下反应式1所示,由易得的结构式ii的(1r,3s)-3-(2,2-二卤乙烯基)-2,2-二甲基-环丙烷羧酸及其盐和酯制备结构式i的(1r,3r)-2,2-二甲基-3-(z)-丙-1-烯-1-基)环丙烷羧酸[i]及其盐和酯的方法。
[0002][0003]
式ii中,r为h、金属、烷基、芳烷基、环烷基,x1=x2=cl、br;
[0004]
式i中,r为h、金属、烷基、芳烷基、环烷基。
背景技术:
[0005]
甲氧苄氟菊酯(metofluthrin)是用于家庭应用中以控制昆虫中携带疾病的媒介的最受欢迎的合成拟除虫菊酯之一。甲氧苄氟菊酯[2,3,5,6-四氟-4-(甲氧基甲基)苄基-(ez)-(1rs,3rs;1rs,3sr)-2,2-二甲基-3-丙-1-烯基环丙烷羧酸盐([2,3,5,6-tetrafluoro-4-(methoxymethyl)benzyl-(ez)-(1rs,3rs;1rs,3sr)-2,2-dimethyl-3-prop-1-enylcyclopropanecarboxylate])以2,3,5,6-四氟-4-(甲氧基甲基)苄基-(z)-(1r,3r)-2,2-二甲基-3-丙-1-烯基环丙烷羧酸盐(2,3,5,6-tetrafluoro-4-(methoxymethyl)benzyl-(z)-(1r,3r)-2,2-dimethyl-3-prop-1-enylcyclopropanecarboxylate)为主要成分[97%以上]组成。
[0006]
(1r,3r)-2,2-二甲基-3-(z)-丙-1-烯-1-基)环丙烷羧酸[i]是制备甲氧苄氟菊酯的重要中间体。
[0007]
美国专利no 6225495描述了使用2,3,5,6-四氟-4-(甲氧基甲基)苄醇iii和衍生自(1r,3r)-2,2-二甲基-3-(2-甲基丙-1-烯基)环丙烷-1-羧酸[菊酸]的酰氯iv,然后臭氧分解所得酯v以提供醛,所述醛通过与乙基三苯基正膦反应提供如方案1中所示的甲氧苄氟菊酯vii。
[0008][0009]
us6225495的方法存在使用昂贵的反应物的问题,还涉及臭氧分解的危险步骤和最后步骤产生废产物的维蒂希(wittig)反应。
[0010]
crombie、christine f.doherty和g.pattenden在j.chem.soc.c,1970,1076-1080中公开的题为“syntheses of 14
c-labelled(+)-trans-chrysanthemum mono-and di-carboxylic acids,and of related compounds”的文章中公开了
14
c标记的(+)-反式-菊花单羧酸和双羧酸的立体特异性合成,其中酸的甲酯是通过(+)-反式-环丙烷醛(viii)和合适的
14
c标记的正膦之间的维蒂希缩合获得。菊花酸甲酯通过醛(viii)转化为菊花二羧酸的总收率为39%。甲基去甲-(9)和双去甲-(8)(
±
)-反式菊酸甲酯是由(viii)与适当的亚烷基正膦缩合而合成的。
[0011]
in191230(35/mas/2001)报道了一种生产结构式i的2,2-二甲基-3-(l-丙烯基)环丙烷羧酸酯的方法,其如方案2所示。
[0012][0013]
in191230的方法存在使用危险的臭氧分解步骤和产生废物的维蒂希反应的问题。而且e、z异构体比例(10:90)也没有达到商业上的要求。
[0014]
美国专利6072074描述了通过如方案3所示的开链前体的环化来合成2,2-二甲基-3-(l-丙烯基)环丙烷羧酸酯。
[0015][0016]
us6072074的方法导致外消旋混合物的形成,其需要在最后步骤中分解,导致至少50%的材料损失。
[0017]
us7393971描述了一种生产2,2-二甲基-3-(l-丙烯基)环丙烷羧酸酯的方法,其中按照下述方案4所描述的,除虫菊酸在喹啉存在下在高温下用氧化铜或铜粉脱羧。
[0018][0019]
方案4中所用的除虫菊酸不易从天然来源获得。
[0020]
美国专利7985872描述了一种制备2,2-二甲基-3-(l-丙烯基)环丙烷羧酸酯i的方法,该方法如方案5所示,通过羟醛缩合得到化合物xvi,并将化合物xvi去甲酰化得到化合物i。
[0021][0022]
us7985872也存在菊酸和臭氧分解反应步骤的可用性差的问题。
[0023]
在bull.chem soc.52,1511-1514(1979)发表的名为“studies on chrysanthemate drivatives.vi.a stereoselective synthesis of trans-3-(2,2-dichloroethenyl)-2,2-dimethyl-1-cyclopropane carboxylic acid and related compounds”的文章描述了反式-3-(2,2-二氯乙烯基)-2,2-二甲基-1-环丙烷羧酸ii的立体选择性合成,该化合物是通过脱氯化氢和使用氢氧化钾在乙醇中水解xx而获得的。在thf中用叔丁醇钾进一步处理ii,得到反式-3-(2-氯乙炔基)-2,2-二甲基-1-环丙烷羧酸乙酯xvii。(方案6)
[0024][0025]
因此,对于开发一种新的且实用的制备作为制备甲氧苄氟菊酯vii的中间体的式i所示的化合物的方法存在着明显的需求。
[0026]
发明目的
[0027]
根据本发明的一个目的,提供了一种以高收率和高纯度制备(1r,3r)-2,2-二甲基-3-(z)-丙-1-烯-1-基)环丙烷羧酸[i](甲氧苄氟菊酯vii的一种高级中间体)的简短而有效的方法。
[0028]
根据本发明的另一个目的,提供了一种以高收率和高纯度制备2,2-二甲基-3-(1-丙烯基)环丙烷羧酸,i(r=h)及其金属盐(r=m+)和酯(r=烷基、芳烷基、环烷基)的简短而有效的方法。
[0029]
根据本发明的另一个目的,提供了一种制备式[i]所示的化合物的方法,该方法避免了使用危险/有毒化学品,也避免了不需要的污染副产物的产生。
[0030]
根据本发明的另一个目的,提供了一种制备式[i]所示的化合物的方法,该方法使本领域技术人员能够更有效和低成本地制备式[i]中间体。
技术实现要素:
[0031]
根据本发明的一种实施方式,提供了一种制备通式i所示的(1r,3r)-2,2-二甲基-3-(z)-丙-1-烯-1-基)环丙烷羧酸及其盐或酯的方法。
[0032][0033]
通式i中,r为h、金属、烷基、芳烷基、环烷基,
[0034]
并如方案7中所示,包括以下步骤:
[0035]
a.使通式ii所示的(1r,3s)-3-(2,2-二卤代乙烯基)-2,2-二甲基-环丙烷羧酸或其盐和酯与强碱在极性溶剂中反应,得到通式xvii所示的(1r,3s)-3-(卤代乙炔基)-2,2-二甲基环丙烷羧酸、盐和酯;
[0036][0037]
通式ii中,r为h、金属、烷基、芳烷基、环烷基,x1=x2=cl、br;
[0038]
r为h、金属、烷基、芳烷基、环烷基,x1=x2=cl、br;
[0039]
b.通过使式xvii所示的(1r,3s)-3-(卤代乙炔基)-2,2-二甲基环丙烷羧酸、盐和酯与丁基锂反应得到乙炔化锂,然后进行步骤(c)中的甲基化步骤,转化通式xvii所示的化合物,得到通式xviii所示的化合物;或者通过在金属盐和配体存在下,使式xvii所示的化合物与甲基格氏试剂反应,得到通式xviii所示的甲基乙炔和少量百分比的通式xix所示的金属乙炔化物的混合物;
[0040][0041]
通式xviii中,r为h、金属、烷基、芳烷基、环烷基;
[0042]
通式xix中,r为h、金属、烷基、芳烷基、环烷基;
[0043]
c.通过向步骤(b)中获得的反应混合物中加入甲基化试剂,将金属乙炔化物xix原位转化为甲基乙炔xviii;
[0044]
d.半氢化/部分还原甲基乙炔xviii中的三键,得到式i所示的化合物,
[0045]
其中,步骤(d)中的半氢化是通过在不同载体上的低百分比非均相催化剂存在下催化加氢或在基于钯、镍、钴、锰、铁或金属组合的催化剂存在下,在氢供体如水、甲酸、甲酸铵、甲酸肼存在下用氢供体转移氢化而实现的。
[0046]
根据本发明的另一种实施方式,甲基乙炔xviii半氢化为通式i所示的化合物是通过以下两种方法中的任意一种实现的:
[0047]
(i)在合适的金属催化剂存在下,通过催化氢化将xviii中的三键部分还原为双键。所得烯烃i主要具有z型几何结构;
[0048]
(ii)或者,通过在合适的催化剂存在下用氢供体转移氢化来半氢化xviii的三键。所得产物i主要是所需的z-异构体。
具体实施方式
[0049]
本发明提供了一种制备通式i所示的(1r,3r)-2,2-二甲基-3-(z)-丙-1-烯-1-基)环丙烷羧酸及其盐或酯的方法,
[0050][0051]
通式i中,r为h、金属、烷基、芳烷基、环烷基,
[0052]
该方法包括以下步骤:
[0053]
a.使通式ii所示的(1r,3s)-3-(2,2-二卤代乙烯基)-2,2-二甲基-环丙烷羧酸或其盐和酯与强碱在极性溶剂中反应,以获得通式xvii所示的(1r,3s)-3-(卤代乙炔基)-2,2-二甲基环丙烷羧酸、盐和酯;
[0054][0055]
通式ii中,r为h、金属、烷基、芳烷基、环烷基,x1=x2=cl、br;
[0056]
通式xvii中,r为h、金属、烷基、芳烷基、环烷基,x1=x2=cl、br;
[0057]
b.通过使式xvii所示的(1r,3s)-3-(卤代乙炔基)-2,2-二甲基环丙烷羧酸、盐和酯与丁基锂反应得到乙炔化锂,然后进行步骤(c)中的甲基化步骤,转化通式xvii的所示的化合物,得到通式xviii所示的化合物;或者通过在金属盐和配体存在下,使式xvii所示的化合物与甲基格氏试剂反应,得到通式xviii所示的甲基乙炔和少量百分比的通式xix所示的金属乙炔化物的混合物;
[0058][0059]
通式xviii中,r为h、金属、烷基、芳烷基、环烷基;
[0060]
通式xix中,r为h、金属、烷基、芳烷基、环烷基;
[0061]
c.通过向步骤(b)中获得的反应混合物中加入甲基化试剂,将金属乙炔化物xix原位转化为甲基乙炔xviii;
[0062]
d.半氢化/部分还原甲基乙炔xviii中的三键,得到式i所示的化合物,
[0063]
其中,步骤(d)中的半氢化是通过在不同载体上的低百分比非均相催化剂存在下
催化加氢或在基于钯、镍、钴、锰、铁或金属组合的催化剂存在下,在氢供体如水、甲酸、甲酸铵、甲酸肼存在下用氢供体转移氢化而实现的。
[0064][0065]
以下将详细描述这些步骤。
[0066]
步骤1
[0067]
在第一步中,将偕二卤代烯基化合物(geminal dihalo alkenyl compound)[式ii;(r=h、金属、烷基、芳烷基、环烷基;x1=x2=cl,br)]在惰性有机溶剂中用强碱处理。优选地,通过用强碱处理,在不影响酸/酯基团的情况下,将3-(2,2-二氯乙烯基)-2,2-二甲基环丙烷羧酸或其酯脱氯化氢成氯代乙炔xvii。
[0068]
所述强碱优选选自例如碱金属和碱土金属氢氧化物或醇盐的碱,例如但不限于氢氧化钾、叔丁醇钾、氢氧化钠和其它强碱,例如氢化钠、叔胺和氨基钠。
[0069]
当r=h,x1=x2=cl时,脱氯化氢在选自极性溶剂但不限于四氢呋喃、二甲基甲酰胺、二甲亚砜、二甲基甲酰胺、n,n-二甲基乙酰胺和n-甲基吡咯烷酮、环丁砜的溶剂中进行。
[0070]
当r=h,x1=x2=cl时,脱氯化氢优选在氢氧化钾的二甲亚砜溶液中进行。
[0071]
碱的用量通常为2.0摩尔当量至3.0摩尔当量。进行反应的温度通常为10-35℃,优选30-35℃,该步骤的反应时间为2-8小时,优选4-5小时。
[0072]
步骤2
[0073]
在金属盐和一些配体存在下,用丁基锂或甲基格氏试剂处理式xvii所示的卤代乙炔,主要得到式xviii所示的甲基乙炔以及一些金属乙炔化物xix。
[0074]
用甲基格氏试剂如甲基卤化镁处理式xvii(r=烷基、芳烷基、环烷基或锂金属)所示的卤代乙炔,其中甲基卤化镁选自甲基碘化镁、甲基溴化镁和甲基氯化镁。
[0075]
所述金属盐选自氯化铜、氯化镍、乙酰丙酮铁,但优选氯化铜。
[0076]
所述配体选自n-甲基吡咯烷(n-methyl pyrrolidine)、n,n-四甲基乙二胺、亚磷酸三乙酯、三苯基膦、三羟甲基膦(tritoluyl phosphine),但优选n-甲基吡咯烷酮(n-methyl pyrrolidone)。
[0077]
这种耦合的实际操作包括使用甲基碘化镁、溴化镁或氯化镁,与氯化铜和n-甲基吡咯烷酮结合。因此,第二步包括在10-35℃,优选20-35℃,最优选25-30℃的温度范围内,在有机惰性溶剂中,用卤化铜(ii)和配体如n-甲基吡咯烷酮处理单卤代炔基化合物(式xvii,r=烷基、芳烷基、环烷基或金属),处理时间为10-90分钟,优选30-90分钟,最优选45-60分钟。
[0078]
卤化铜(ii)可以是氯化铜(ii)、溴化铜(ii)、碘化铜(ii),最优选氯化铜(ii)。卤化铜(ii)的量可以为2摩尔%-50摩尔%,优选5摩尔%-15摩尔%,最优选4摩尔%-8摩尔%。
[0079]
配体的量可以为2摩尔%-20摩尔%,优选5-10摩尔%。
[0080]
步骤(b)中的反应在选自乙醚、四氢呋喃、二恶烷(dioxane)或苯甲醚的溶剂中单独进行,或在与烃溶剂如己烷、甲苯、二甲苯的混合溶剂中进行。
[0081]
反应的下一部分是使该混合物与甲基卤化镁在相同的惰性溶剂中反应。所述甲基卤化镁可以是甲基氯化镁、甲基溴化镁或甲基碘化镁。甲基卤化镁的加入时间可以是30分钟-180分钟,优选60-120,最优选60-90分钟。
[0082]
反应温度可以为0-25℃,最优选0-10℃,甲基卤化镁的量可以是1.0-2.0摩尔当量,最优选1.2-1.5摩尔当量。
[0083]
步骤3
[0084]
通过向反应混合物中原位加入甲基化试剂,例如甲基碘、甲基氯、甲基溴、碳酸二甲酯和硫酸二甲酯,优选碳酸二甲酯,将金属乙炔化物xix转化为甲基乙炔xviii。甲基化试剂的量可以在0.1-0.5摩尔当量,最优选0.2-0.3摩尔当量的范围内变化。
[0085]
然后使反应混合物与甲基化试剂在与前述相同的温度下反应。0-25℃,最优选0-10℃。
[0086]
当底物是式xvii所示的羧酸(r=h,x=cl)时,将其转化为金属羧酸盐,以节省该
步骤中使用的甲基卤化镁的量。在惰性有机溶剂中,用碱金属碱处理式xvii(r=h,x=cl)所示的羧酸。
[0087]
所述碱金属碱可以是氢氧化钠、氢氧化钾、氢氧化锂、碳酸锂、甲醇锂,或碱土金属氢氧化物如氢氧化镁和氢氧化钙,或碱土金属氧化物/醇盐如氧化镁、甲醇镁。
[0088]
所述惰性溶剂可以是非极性溶剂,例如甲苯、二甲苯、丙酮、甲基异丁基酮、四氢呋喃、二氯甲烷、氯仿、甲基叔丁基醚(mtbe)。
[0089]
所述反应的温度可以为110-140℃,任选在氮气压下。
[0090]
将得到的水或甲醇与惰性溶剂一起蒸馏除去。
[0091]
然后用惰性溶剂稀释金属羧酸盐并用于下一步骤。所用的惰性溶剂为四氢呋喃。惰性溶剂的用量通常为10-30重量份,优选10重量份。无机碱的用量通常为1.0-1.5摩尔当量,最优选1.05摩尔当量。
[0092]
步骤4
[0093]
甲基乙炔xviii(r=h、烷基、芳烷基或环烷基)的半氢化提供了所需的式i所示的(1r,3r)-2,2-二甲基-3-(z)-丙-1-烯-1-基)环丙烷羧酸或其酯,其中r=h、烷基、芳烷基或环烷基。式xviii所示的甲基乙炔的半氢化通过(i)在合适催化剂存在下的催化氢化(ii)在合适催化剂存在下与氢供体进行半氢化来实现。
[0094]
乙炔的半氢化通常在lindlar催化剂存在下进行[us 6072074],尽管它具有以下缺点:
[0095]
a)它含有5%乙酸铅中毒的钯,而低百分比的钯催化剂可以在不使用铅中毒的情况下实现相同的转化率。
[0096]
b)使用lindlar催化剂的氢化的立体选择性较差,需要通过加入二级毒物如喹啉来改善。
[0097]
本发明的发明人发现,以下两种方法在将式xviii所示的化合物转化为所需的式i所示的z-烯烃方面得到了良好的结果:
[0098]
(i)在合适的金属催化剂存在下,xviii所示的化合物中的三键通过催化加氢作用被部分还原为双键。
[0099]
(ii)或者,在合适的催化剂存在下,通过用氢供体转移氢化将xviii所示的化合物中的三键半氢化。
[0100]
所述半氢化通过在不同载体上的低百分比非均相钯催化剂的存在下的催化加氢,或在钯或其它混合金属催化剂和氢供体的存在下进行转移氢化来实现。
[0101]
甲基乙炔xviii转化为所需的式i所示的z-烯烃优选通过如下方式完成:
[0102]
a)在负载0.5%钯的碳或二氧化钛存在下,氢化甲基乙炔xviii;
[0103]
b)甲基乙炔xviii在二甲基甲酰胺和水中,在乙酸钯存在下通过转移氢化作用而被还原。
[0104]
式xviii所示的甲基乙炔的半氢化是通过在不同载体上的低百分比非均相催化剂的存在下催化加氢,或在基于钯、镍、钴、锰、铁或金属组合的催化剂的存在下,在氢供体如水、甲酸、甲酸铵、甲酸肼的存在下,用氢供体进行转移氢化来实现的。
[0105]
式xviii所示的甲基乙炔的半氢化优选在负载0.5-1.0%钯的碳或二氧化钛和气态氢的存在下,在选自烃如己烷或醇如甲醇、乙醇、异丙醇、2-甲氧基乙醇等的溶剂中进行。
在步骤(d)中,式(viii)的半氢化/部分还原而形成式(i)所示的化合物优选通过在乙酸钯和碱如氢氧化钠、氢氧化锂、氢氧化钾或碱土金属氢氧化物如氢氧化镁和合适的溶剂存在下进行转移氢化而实现,所述合适的溶剂选自二甲基甲酰胺、水和醇如甲醇、乙醇。
[0106]
所述转移氢化在选自醇如甲醇、乙醇或极性溶剂如二甲基甲酰胺的合适的溶剂中进行。反应在50-150℃,优选130-150℃的高温下进行。
[0107]
以下实施例说明了制备式i所示的化合物的方法步骤,但本发明并不限于此。
[0108]
1.使用强碱由(1r,3s)-3-(2,2-二氯乙烯基)-2,2-二甲基环丙烷羧酸(ii)制备(1r,3s)-3-(氯乙炔基)-2,2-二甲基环丙烷羧酸(xvii)。
[0109]
向二甲基亚砜(30ml)和(1r,3s)-3-(2,2-二氯乙烯基)-2,2-二甲基环丙烷羧酸(10g,0.047摩尔)的混合物中;加入氢氧化钾(5.33g,0.095摩尔),并在室温下搅拌4小时。用水淬灭反应混合物,并用稀盐酸水溶液将反应混合物的ph调节至酸性。将所得产物萃取到甲苯中。在真空下蒸馏甲苯层,得到浅黄色油状的(1r,3s)-3-(氯乙炔基)-2,2-二甲基环丙烷羧酸,产率为97%,gc纯度为99.8%。
[0110]
h1nmr(cdcl3,内标-tms,400mhz),δ值(ppm):1.27(s,3h),1.32(s,3h),1.70(d,1h),1.96(dd,1h)。
[0111]
2.使用强碱由(1r,3s)-3-(2,2-二氯乙烯基)-2,2-二甲基环丙烷羧酸叔丁酯(ii)制备(1r,3s)-3-(氯乙炔基)-2,2-二甲基环丙烷羧酸叔丁酯(xvii)。
[0112]
向二甲基亚砜(30ml)和(1r,3s)-3-(2,2-二氯乙烯基)-2,2-二甲基环丙烷羧酸叔丁酯(10g,0.037摩尔)的混合物中;加入氢氧化钾(4.22g,0.075摩尔)并在室温下搅拌4小时。用水淬灭反应混合物,并用稀盐酸水溶液将反应混合物的ph调节至酸性。将所得产物萃取到甲苯中。在真空下蒸馏甲苯层,得到浅黄色油状的(1r,3s)-3-(氯乙炔基)-2,2-二甲基环丙烷羧酸叔丁酯,产率为97%,gc纯度为99.8%。
[0113]h1 nmr(cdcl3,内标-tms,400mhz),δ值(ppm):1.20(s,3h),1.27(s,3h),1.44(s,9h),1.61(d,1h),1.84(dd,1h)。
[0114]
3.由(1r,3s)-3-(氯乙炔基)-2,2-二甲基环丙烷羧酸(xvii)制备(1r,3r)-2,2-二甲基-3-(丙-1-炔-1-基)环丙烷羧酸(xviii)。
[0115]
(1r,3s)-3-(氯乙炔基)-2,2-二甲基环丙烷羧酸(10g,0.058摩尔)、lioh
·
h2o氢氧化锂一水合物(2.45g,0.058摩尔)和甲苯(200ml)加热至110-112℃,通过dean-stark装置将甲苯和水的共沸物完全蒸出。用thf(100ml)稀释所得固体。将cucl2(4摩尔%)和n-甲基吡咯烷酮(5摩尔%)加入到反应混合物中,并在室温下搅拌60分钟。将反应混合物冷却至0-5℃,在0-5℃下,在60分钟内,在搅拌条件下向其中加入甲基碘化镁(3.0m,溶于24ml二乙醚中,0.071摩尔),并在0-5℃下再搅拌60分钟。将甲基碘(2.1g,0.014摩尔)加入到反应混合物中并在室温下搅拌15小时。向反应混合物中加入水并搅拌3分钟。分离各层,水相用稀盐酸溶液酸化,然后向反应物料中加入甲苯。在真空下蒸馏甲苯层,得到粗品(1r,3r)-2,2-二甲基-3-(丙-1-炔-1-基)环丙烷羧酸,为棕色粘稠油状物,产率为85%,gc纯度为95%。
[0116]
h1nmr(cdcl3,内标-tms,400mhz),δ值(ppm):1.24(s,3h)、1.27(s,3h)、1.60(d,1h)、1.79(d,3h)、1.90(dd,1h)。
[0117]
4.由(1r,3s)-3-(氯乙炔基)-2,2-二甲基环丙烷羧酸乙酯(xvii)制备(1r,3r)-2,2-二甲基-3-(丙-1-炔-1-基)环丙烷羧酸乙酯(xviii)。
[0118]
(1r,3s)-3-(氯乙炔基)-2,2-二甲基环丙烷羧酸乙酯(10g,0.05摩尔)、lioh
·
h2o氢氧化锂一水合物(2.1g,0.05摩尔)和甲苯(200ml)的混合物加热至110-112℃,通过dean-stark装置将甲苯和水的共沸物完全蒸出。用thf(100ml)稀释所得固体。将cucl2(4摩尔%)和n-甲基吡咯烷酮(5摩尔%)加入到反应混合物中,并在室温下搅拌60分钟。将反应混合物冷却至0-5℃,在0-5℃下,于60分钟内,在搅拌条件下向其中加入甲基碘化镁(3.0m,溶于20ml二乙醚中,0.06摩尔),将反应混合物在0-5℃下搅拌60分钟。将甲基碘(2.1g,0.014摩尔)加入到反应混合物中并在室温下搅拌15小时。向反应混合物中加入水并搅拌3分钟。分离各层,水相用稀盐酸溶液酸化,然后加入到反应物料中。在真空下蒸馏甲苯层,得到粗品(1r,3r)-2,2-二甲基-3-(丙-1-炔-1-基)环丙烷羧酸乙酯,为棕色粘稠油状物,产率为85%,gc纯度为95%。
[0119]
h1nmr(cdcl3,内标-tms,400mhz),δ值(ppm):1.12(s,3h),1.22(s,6h),1.57(d,1h),1.73(s,3h),1.85(d,1h),4.11(q,2h)
[0120]
5.由(1r,3s)-3-(氯乙炔基)-2,2-二甲基环丙烷羧酸叔丁酯(xvii)制备(1r,3r)-2,2-二甲基-3-(丙-1-炔-1-基)环丙烷羧酸叔丁酯(xviii)。
[0121]
将(1r,3s)-3-(氯乙炔基)-2,2-二甲基环丙烷羧酸叔丁酯(10g,0.043摩尔)、lioh
·
h2o氢氧化锂一水合物(1.83g,0.043摩尔)和甲苯(200ml)的混合物加热至110-112℃,通过dean-stark装置将甲苯和水的共沸物完全蒸出。用thf(100ml)稀释所得固体。将cucl2(4摩尔%)和n-甲基吡咯烷酮(7摩尔%)加入到反应混合物中,并在室温下搅拌60分钟。将反应混合物冷却至0-5℃,在搅拌条件下于60分钟内于0-5℃向其中加入甲基碘化镁(3.0m,溶于17ml二乙醚中,0.051摩尔),将反应混合物于0-5℃再搅拌60分钟。将甲基碘(2.1g,0.014摩尔)加入到反应混合物中并在室温下搅拌15小时。向反应混合物中加入水(200ml)并搅拌3分钟。分离各层,水相用稀盐酸溶液酸化,然后向反应物料中加入甲苯。在真空下蒸馏甲苯层,得到粗品(1r,3r)-2,2-二甲基-3-(丙-1-炔-1-基)环丙烷羧酸叔丁酯,为棕色粘稠油状物,产率为85%,gc纯度为95%。
[0122]
h1nmr(cdcl3,内标-tms,400mhz),δ值(ppm):1.20(s,3h)、1.26(s,3h)、1.44(s,9h)、1.52(d,1h)、1.80(d,1h)、1.81(s,3h)。
[0123]
6.使用乙酸钯与二甲基甲酰胺/koh选择性半氢化(1r,3r)-2,2-二甲基-3-(丙-1-炔-1-基)环丙烷羧酸(xviii)。
[0124]
将(1r,3r)-2,2-二甲基-3-(丙-1-炔-1-基)环丙烷羧酸(0.76g,0.0050摩尔)、二甲基甲酰胺(10ml)、koh(0.42g,0.0075摩尔)和乙酸钯(0.0245g,0.000109摩尔)的混合物在密封管中在145℃下加热并在该温度下保持4小时。将反应混合物冷却至室温。过滤催化剂并用二甲基甲酰胺洗涤。向滤液母液中加入水(10ml)和甲苯(10ml),并在室温下搅拌。分离各层,水层用稀盐酸溶液酸化,产物用甲苯(10ml)萃取。在真空下蒸馏甲苯层,得到0.72g(1r,3r)-2,2-二甲基-3-[(1z)-丙-1-烯-1-基]环丙烷羧酸,z/e异构体的比例为97.5/2.5。
[0125]
h1nmr(cdcl3,内标-tms,400mhz),δ值(ppm):1.20(s,3h)、1.32(s,3h)、1.47(d,1h)、1.71(d,3h)、2.19(d,1h)、5.15(d,1h)、5.61(d,1h)。
[0126]
7.使用乙酸钯与二甲基甲酰胺/koh选择性半氢化(1r,3r)-2,2-二甲基-3-(丙-1-炔-1-基)环丙烷羧酸乙酯(xviii)。
[0127]
将(1r,3r)-2,2-二甲基-3-(丙-1-炔-1-基)环丙烷羧酸乙酯(0.90g,0.0050摩
尔)、二甲基甲酰胺(10ml)、koh(0.42g,0.0075摩尔)和乙酸钯(0.0245g,0.000109摩尔)的混合物在密封管中在145℃下加热并在相同温度下保持4小时。将反应混合物冷却至室温。过滤催化剂并用二甲基甲酰胺洗涤。向滤液中加入水和甲苯,并在室温下搅拌。分离各层,水层用稀盐酸溶液酸化,产物萃取到甲苯中。在真空下蒸馏甲苯层,得到0.85g(93.4%)(1r,3r)-2,2-二甲基-3-[(1z)-丙-1-烯-1-基]环丙烷羧酸乙酯,其z/e异构体比例为97.5/2.5。
[0128]
(通过气液色谱法测得)
[0129]
h1nmr(cdcl3,内标-tms,400mhz),δ值(ppm):1.20(s,3h)、1.32(s,3h)、1.47(d,1h)、1.71(d,3h)、2.19(d,1h)、5.15(d,1h)、5.61(d,1h)。
[0130]
8.使用乙酸钯与二甲基甲酰胺/koh选择性半氢化(1r,3r)-2,2-二甲基-3-(丙-1-炔-1-基)环丙烷羧酸叔丁酯(xviii)。
[0131]
将(1r,3r)-2,2-二甲基-3-(丙-1-炔-1-基)环丙烷羧酸叔丁酯(1.04g,0.0050摩尔)、二甲基甲酰胺(10ml)、koh(0.42g,0.0075摩尔)和乙酸钯(0.0245g,0.000109摩尔)的混合物在密封管中在145℃下加热并在该温度下保持4小时。将反应混合物冷却至室温。过滤催化剂并用二甲基甲酰胺洗涤。向滤液中加入水和甲苯,并在室温下搅拌。分离各层,水层用稀盐酸溶液酸化,产物萃取到甲苯中。在真空下蒸馏甲苯层,得到0.95g(93.4%)(1r,3r)-2,2-二甲基-3-[(1z)-丙-1-烯-1-基]环丙烷羧酸叔丁酯,其z/e异构体比例为97.5/2.5。(通过气液色谱法测得)
[0132]
9a.使用负载0.5%钯的碳选择性半氢化(1r,3r)-2,2-二甲基-3-(丙-1-炔-1-基)环丙烷羧酸乙酯(xviii)
[0133]
加入(1r,3r)-2,2-二甲基-3-(丙-1-炔-1-基)环丙烷羧酸乙酯(0.90g,0.0050摩尔)、己烷(10ml)、负载0.5%钯的碳(0.45g)的混合物。氮气吹扫30分钟,并通过氢气吹扫抽空。氢气被吹至进气口,停止使用。过滤催化剂并在氮气气氛下用己烷洗涤。真空蒸馏己烷层,得到0.85g(1r,3r)-2,2-二甲基-3-[(1z)-丙-1-烯-1-基]环丙烷羧酸乙酯,z/e异构体的比例为98/2.0。
[0134]
h1nmr(cdcl3,内标-tms,400mhz),δ值(ppm):1.20(s,3h)、1.32(s,3h)、1.47(d,1h)、1.71(d,3h)、2.19(d,1h)、5.15(d,1h)、5.61(d,1h)。
[0135]
9b.使用负载0.5%钯的硅酸钛选择性半氢化(1r,3r)-2,2-二甲基-3-(丙-1-炔-1-基)环丙烷羧酸乙酯(xviii)
[0136]
采用负载0.5%钯的硫酸钛代替负载0.5%钯的碳,重复9a中的方法。最终产物为0.85g的(1r,3r)-2,2-二甲基-3-[(1z)-丙-1-烯-1-基]环丙烷羧酸乙酯,z/e异构体比例为98.5/1.5。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1