用于制备生物活性共聚物的工艺的制作方法
1.本发明涉及用于制备生物活性共聚物的工艺,该生物活性共聚物包含衍生自丙烯醛单体的链段和聚亚烷基二醇链段。本发明进一步涉及生物活性试剂在治疗疾病,特别是在治疗细菌感染、病毒感染或癌症中的用途。
背景技术:
2.存在日益增长的对于有效的抗微生物、抗病毒和抗癌症试剂的需要。病原体的抗微生物剂耐受性的演进已导致严重的健康风险,这是由于通过许多常规抗微生物剂不能够控制感染。新型病毒株系的出现导致需要广谱治疗,特别是对于严重的病毒感染。在2020年期间,由sars-cov-2引起的全球流行病的出现已经导致全世界数以百万计的感染,在老年人和患有并发症的人中大量的死亡。这已导致迫切需要有效地治疗sars-cov-2感染。
3.还迫切需要有效地治疗细菌感染,特别是严重和威胁生命的细菌感染,诸如败血症(sepsis)。2017年,败血症的全球发生率和死亡率估计为4890万人,其中1100万与败血症相关的死亡(the lancet vol 395,issue 10219 pp200-211(2020年1月18日)。以抗微生物剂耐受性为目标的干预对于改进败血症结果是急迫的。
4.我们的共同未决的专利申请wo2016/077879和wo2017/139849公开了抗微生物共聚物,该抗微生物共聚物包含衍生自丙烯醛单体的链段和聚亚烷基二醇链段。共聚物通过在25℃至35℃的温度下使聚丙烯醛与聚亚烷基二醇反应而形成,以提供具有针对宽范围的细菌和病毒的良好活性的抗微生物剂。聚合物型抗生素呈现出在治疗感染(诸如败血症)方面的显著进步,这是由于它们的活性,以及由于共聚物的作用模式而使细菌和病毒株系发展出耐受性的倾向降低。
5.一直需要提供具有改进的活性、特别是抗微生物活性的生物活性化合物。
技术实现要素:
6.我们现已发现,若包含丙烯醛衍生的链段和聚亚烷基二醇低聚物的聚合物是在低温下形成的,则该共聚物的生物活性显著增加。因此,我们提供用于制备包含丙烯醛衍生的链段和聚亚烷基二醇低聚物链段的生物活性聚合物的工艺,该工艺包含在不大于15℃、优选地不大于12℃、诸如不大于10℃或不大于8℃的温度使聚亚烷基二醇与丙烯醛在水溶液中反应,以形成分子量不大于1000道尔顿的共聚物。
7.在进一步实施方式中,提供治疗患有选自微生物感染、病毒感染和癌症的疾病的受试者的方法,该方法包含对受试者给药有效量的根据所述工艺制备的生物活性试剂。
8.还提供丙烯醛和聚亚烷基二醇在制造用于治疗选自细菌感染,病毒感染和癌症的疾病的药剂中的用途,其中该用途包含所述工艺。
9.所述工艺特别适用于治疗癌症、细菌感染或病毒感染。
10.我们已经发现,在低温下形成的共聚物的活性非常显著地增加。事实上,最小抑制浓度(mic),阻碍细菌(一种或多种细菌)的可见生长的最低浓度,在较低温度下非常显著地
更低。例如,mic在40℃下一般是在10℃下的至少三倍高。事实上,对于很多细菌而言,mic在40℃下是在10℃下的四倍高和甚至至少六倍高。
附图说明
11.参考附图描述本发明的实例。在附图中:
12.图1是具有实施例2中提到的四个线图的图,其比较了使用如下治疗的烧伤伤口感染模型的菌落形成单元计数(log
10
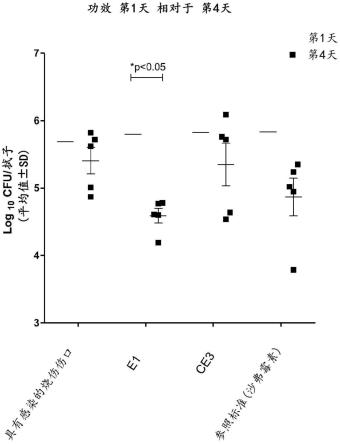
cfu/拭子):(i)本发明工艺的共聚物组合物(e1),(ii)非本发明的对比工艺的共聚物组合物(ce3),(iii)使用已知抗微生物剂(沙弗霉素(safromycin)),和(iv)未治疗的对照。
13.图2是具有实施例2中提到的四个线图的图,其比较了在使用图1中提到的组合物治疗后,感染四天之后的伤口收缩。
14.图3是具有实施例3中提到的五个线图的图,其比较了在使用如下四种治疗中的每一种来治疗上行尿道感染后,肾组织的菌落形成单元计数(log
10
cfu/g):以两种不同处理速率(治疗速率)的本发明工艺的共聚物组合物(e1);使用抗生素美罗培南(meropenem)的阳性对照,以及早期和晚期感染对照。
15.图4是显示实施例3中提到的五个线图的图,其比较了在使用如下四种治疗中的每一种来治疗上行尿道感染后,膀胱组织的菌落形成单元计数(log
10
cfu/g):以两种不同的处理速率的本发明工艺的共聚物组合物(e1);使用抗生素美罗培南的阳性对照,以及早期和晚期感染对照。
16.图5是显示仓鼠中鼻洗剂滴度(log
10
染色体组s/μl)的图,该仓鼠被sars-cov-2感染,并且使用组合物e1和ce3治疗,如实施例6中所描述。
17.图6是显示如下的柱状图:在实施例7中提到的使用由人类呼吸道上皮细胞制得的类器官的体外研究中,sars-cov-2病毒的浓度依赖性降低。
18.图7是显示如下的柱状图:共聚物e1在实施例7中提到的不同浓度(ppm)下在vero细胞方案中观察到的最大细胞毒性百分数。
19.图8是显示如下的柱状图:共聚物ce1在实施例7中提到的不同浓度(ppm)下在vero细胞方案中观察到的最大细胞毒性百分数。
20.图9是显示在实施例8中提到的使用参照物以及实施例e1和ce3的组合物治疗后的阴道拭子中的细菌载量的图。
具体实施方式
21.术语“身体”意指人和/或动物的身体;术语“受试者(subject)”意指这样的身体,其为所述受试者。
22.静脉疗法(简称iv疗法或iv疗法)是将液体物质直接地输注到静脉中。
23.如本文中所用的,术语“肠胃外(parenteral)”意指以不同于通过完整消化道的方式进入(摄入)身体内。也就是说,不在正常的胃或肠内;非肠内的。通过所述工艺制备的共聚物的肠胃外给药是优选的。
24.术语“肠胃外感染”意指不是通过在胃肠道内的方式进入身体而染上的感染。这样的感染可如下发生:经由血管(血液/淋巴)系统,生殖-泌尿道,由肺,皮肤或外保护膜的破
裂,诸如在手术中、针刺伤、割伤、擦伤、或者任何皮肤中的或在皮肤和粘膜之间的间隙中的破坏。将理解,在肠胃外感染和药品的肠胃外给药之间进行清晰的区分,肠胃外感染可潜在地经由包括口服给药在内的任何药品给药方法治疗(假设有效剂量到达感染位点(部位,site)),药品的肠胃外给药限于不同于口服方式的给药。
25.如在本文中提到细菌病原体时使用的,术语“抗生素耐受的”或“超级细菌(superbug)”指的是这样的细菌病原体:其可经受本领域中用于处理该细菌病原体(即,该细菌病原体的非耐受性株系)的抗生素。例如,金黄色葡萄球菌(staphylococcus aureus)可使用甲氧西林(methicillin)处理;然而金黄色葡萄球菌的抗生素耐受的株系,金黄色葡萄球菌usa 300,是耐受甲氧西林的金黄色葡萄球菌(mrsa)。虽然该细菌株系是常见的,但是黄色葡萄球菌:usa:300典型地感染那些免疫受损的或处在易感环境(susceptible environment)中的人。感染将通常通过小割伤或溃疡而进入身体。其它与usa:300相关联的症状为肺炎,引起坏死的筋膜炎,心内膜炎,以及骨头和关节感染。
26.术语“肺给药(肺施用,pulmonary administration)”指的是通过吸入(吸气,inhailation)将本发明的制剂给药至肺中。
27.术语“聚亚烷基二醇”包括c2至c4亚烷基二醇单元的线型和支化的聚醚均聚物和共聚物。优选的聚亚烷基二醇为聚乙二醇和聚丙二醇及其共聚物,并且最优选的是聚乙二醇。
28.术语“全身的(systemic,系统的)”指的是距原始感染位点有距离的或者涉及有机体的整个身体(全身)的疾病或障碍(disorder,紊乱)或原始受伤位点。因此,术语“局部”在本文中是关于原始感染的位点而使用的。因而,全身感染是这样的感染,在该种感染中,病原体存在于器官或血液中(包括菌血症),并且可与严重的、潜在地威胁生命的疾病诸如败血症相关联。局部感染是这样的感染,在该感染中,病原体最远仅迁移至感染的局部组织,诸如肺或伤口的位点。
29.如本文中使用的,术语“吸入(inhalation)”指的是将空气摄入至肺的肺泡中。在具体实例中,摄入可通过在吸入的同时本发明的制剂的自给药,或者通过经由呼吸器给药(例如给药至呼吸器上的患者)而发生。关于本发明的制剂所使用的术语“吸入”与“肺给药”同义。
30.术语“治疗(treatment,处理)”和“治疗(treating,处理)”旨在亦涵盖预防(prophylaxis)、疗法(therapy)和痊愈(治愈,cure)。因此,在一方面中,治疗涉及预防或延迟或延缓与抗生素耐受的细菌相关联的病症(condition)、疾病、或障碍(例如,与疾病、病症、或障碍相关联的症状)的发作。在另一方面中,治疗涉及治疗(例如,最小化或减少或延缓进行或倒退)与抗生素耐受的细菌相关联的现有的病症、疾病、或障碍(例如与疾病、病症、或障碍相关联的症状)。在一个实施方式中,治疗为病症、疾病、或障碍提供痊愈。
31.如本文中使用的短语“药学上可接受的载体”是指在将主题共聚物和/或组合物从一个器官或身体的一部分携带或运输到另一器官或身体的一部分中涉及的药学上可接受的材料、组合物或载剂(vehicle),例如液体或固体填充剂、稀释剂、赋形剂(excipient)或溶剂包封(胶囊)材料。各载体必须在与制剂的其它成分相容的意义上是“可接受的”,并且不对患者造成不适当的伤害。可用作药学上可接受的载体的材料的一些实例包括:糖,如乳糖、葡萄糖和蔗糖;淀粉,如玉米淀粉和马铃薯淀粉;纤维素及其衍生物,如羧甲基纤维素钠、乙基纤维素和乙酸纤维素;粉状黄蓍胶;麦芽;明胶;滑石;赋形剂,如可可脂和栓剂蜡;
油,如花生油、棉籽油、红花油、芝麻油、橄榄油、玉米油和大豆油;二醇,如丙二醇;多元醇,如甘油、山梨醇、甘露醇和聚乙二醇;酯,如油酸乙酯和月桂酸乙酯;琼脂;缓冲剂,如氢氧化镁和氢氧化铝;海藻酸;无热原水;等渗盐水;林格氏(ringer)溶液;乙醇;ph缓冲溶液;聚酯、聚碳酸酯和/或聚酐;和药物制剂中使用的其它无毒相容物质。
32.术语丙烯醛衍生的链段指的是包含一个或多个丙烯醛单体残基的共聚物段。
33.术语低聚物、聚亚烷基二醇低聚物和聚丙烯醛低聚物指的是由至少两个单体单元、优选地至少三个单体单元组成的聚合物。低聚物将典型地包含2至20个单体单元;在一个实施方式中,单元的数量为2至10。
34.术语“单体单元”和“单体残基”是指存在于由反应单体例如丙烯醛和聚亚烷基二醇衍生的(得到的)共聚物中的单元。聚亚烷基二醇视为一般提供至少三个二醇单体单元的预聚物。
35.多分散指数是聚合物的重均分子量(mw)对聚合物的数均分子量(mn)的比率。聚合物的重均分子量和数均分子量可通过分析方法(诸如高效液相色谱)确定。一旦确定了重均分子量和数均分子量,通过将重均分子量除以数均分子量mw/mn可容易地计算多分散指数。假定单分散的聚合物具有1.000的多分散指数。然而,典型的商业聚合物诸如可商购获得的树脂具有10或更多的多分散指数。具有宽分子量分布的聚合物具有较高的多分散指数,并且具有窄分子量分布的聚合物具有较低的多分散指数。
36.术语“间接热交换”意指在没有流体彼此之间的任何物理接触或混杂(intermix)的情况下使流体进入热交换关系。冷却流体在聚合工艺外部。
37.在整个说明书中,使用术语“包括”或“包含”或关于其的语法变体应视为具体说明了存在所述的特征、整数、步骤或组分,但是不排除存在或添加一个或多个未具体提及的其它特征、整数、步骤、组分或其集合。
38.用于制备生物活性物的工艺包含:在不大于15℃、优选地不大于12℃、诸如不大于10℃或不大于8℃的温度下,使聚亚烷基二醇与丙烯醛在水溶液中反应,以形成分子量不大于1000道尔顿的共聚物。
39.在不希望受制于理论的情况下,据信在低温下增强了具有疏水悬垂基团(侧链基团)的更具活性的共聚物的形成。聚合物内的官能团和相应的目标细菌、病毒或癌症(肿瘤,cancer)之间的反应一般通过其之间的疏水吸引而增强。疏水吸引以gibbs自由能度量,其随着焓减少或熵增加而发生(如以gibbs热力学关系δg=δh-tδs的等式所示),其尤其是聚合物中的疏水的乙烯基或亚烷基基团与目标物(细菌、病毒或癌症)中的疏水蛋白之间的许多相互作用的结果。
40.丙烯醛单体(2-丙烯-酮)可与亚烷基二醇低聚物反应,或者通过疏水的乙烯基使链增长并留下羰基悬垂,或者通过羰基使链增长并留下疏水的乙烯基悬垂且未受阻。这两种替代方案中的后者具有悬垂的疏水乙烯基,增强了疏水机理。
41.在不希望受制于理论的情况下,据信通过丙烯醛单体的羰基基团而不是通过丙烯醛单体的乙烯基基团的聚合的发生在不大于15℃、特别地不大于12℃、诸如不大于10℃、或不大于8℃的低温下更为显著,因为羰基聚合的驱动力(焓减少)较弱,以及结果在较高温度下解聚。
42.本领域技术人员将意识到,本构成单体的类似物(analogue,同系物)可合成为交
替或嵌段构型,从而以由其相应的单体型反应性比率支配的方式影响亲水性和从而的生物反应性。
43.所述工艺典型地涉及在碱性催化的条件下使聚亚烷基二醇与丙烯醛在水溶液中反应。优选地,ph不大于12.5,并且优选地在大于7.0至12.5、诸如8至12.0的ph范围内。
44.典型地,聚亚烷基二醇和丙烯醛的水溶液以如下的量包含水:至少20%w/w,诸如20%w/w至80%w/w、20%w/w至70%w/w,或20%w/w至60%w/w。
45.聚亚烷基二醇:丙烯醛的重量比典型地为至少4:1、优选地至少5:1。例如该重量比可为约4:1至20:1,诸如5:1至20:1,或10:1至15:1。
46.共聚物的分子量为不大于1000道尔顿。共聚物可具有250至1000道尔顿、特别地300至1000道尔顿、诸如400至1000道尔顿的分子量。在所述工艺中,聚亚烷基二醇典型地具有不大于600道尔顿、诸如200至600道尔顿的分子量。
47.所述工艺可方便地通过向聚亚烷基二醇的水溶液添加丙烯醛而进行。聚乙二醇的水溶液可包含至少20%w/w水。可控制聚丙烯醛向含水聚乙二醇的添加以使放热反应对增加溶液温度的影响最小化。可使用本领域中已知的一系列方法在丙烯醛单体和聚乙二醇的反应期间控制温度。可使水溶液与热交换器接触,以在添加丙烯醛单体期间提供反应混合物的冷却。例如,可使用计算机化的(computerized)反应器系统精确地控制温度,以基于在所需温度下开始而维持含水组合物的温度。
48.由于反应是放热的,所述工艺优选地包括从丙烯醛单体和聚亚烷基二醇的反应区移去反应热。移去热可通过如下实现:向反应区中添加另外的溶剂(诸如另外的水)或反应物,或者经由可商购获得的和熟知的热交换装置(诸如壳和管或螺旋缠绕热交换器)而将热从反应区转移,所述热交换装置设置有冷却用流体流诸如水流。
49.在一个实施方式中,可通过如下控制丙烯醛和聚亚烷基二醇的放热反应的影响并且在制备共聚物期间将温度维持在所需的范围中:使聚合反应在设置有间接热交换的反应容器中进行以维持所需的温度。在一个实施方式中,反应容器为带夹套的反应容器,并且通过在夹套中提供冷却用流体(诸如水)流来提供间接热交换。该工艺可包括通过调整冷却用流体流和/或添加反应物(诸如丙烯醛单体)的速率而维持温度在不大于15℃、优选地不大于12℃。反应可在合适的反应器中进行,并且在不大于15℃,优选地不大于12℃、诸如不大于10℃、或不大于8℃的所需温度下开始,并且该温度维持在所需范围中。
50.在优选实施方式中,可通过在围绕反应器的至少一部分的夹套中的冷却介质(诸如水或其它冷却剂流体,取决于所需的温度),使用间接交换将聚合热从反应器移去。可计算机控制热移去的效率,以在共聚物的形成期间维持温度在所需范围内。反应器可为具有夹套(用于冷却用流体流)的间歇反应器,或者可为管式反应器,诸如包含一个或多个同心围绕管式反应器的至少一部分的夹套(用于冷却用流体,诸如水)的管式环管反应器。
51.典型地,反应容器将包括搅动器,诸如搅拌器或提供混合的其它手段,以使作为反应放热和/或热交换的结果的相对热或冷的区域的出现最小化。
52.所述工艺可在一组实施方式中以在-20℃至15℃、优选地不大于12℃、诸如-10℃至12℃、或-10℃至10℃范围内的温度实施。典型地,反应物溶液的温度将为至少-10℃。
53.在进一步的一组实施方式中,温度在-10℃至15℃、诸如-5℃至15℃、或0℃至15℃的范围内。在进一步的一组实施方式中,温度在-5℃至12℃、或-5℃至10℃的范围内。
54.在一组实施方式中,水溶液的温度维持在不大于10℃、诸如不大于5℃。
55.所述工艺优选地涉及向聚亚烷基二醇的水溶液添加丙烯醛。可向包含至少20%w/w水的聚乙二醇水溶液添加丙烯醛,其中丙烯醛是以具有不大于50%w/w、优选地不大于30%w/w丙烯醛单体的浓度的丙烯醛水溶液的形式添加的。
56.聚亚烷基二醇可为包含乙二醇、丙二醇、和丁二醇单体中的一种或多种的共聚物,当存在多于一种单体时,该共聚物可以无规或嵌段分布包含单体单元。更优选的聚亚烷基二醇是分子量200至600道尔顿、诸如200至400道尔顿的聚乙二醇。本领域技术人员将理解,术语聚乙二醇优选地不包括一缩二乙二醇。平均分子量200至600道尔顿的聚乙二醇包括标称分子量200至600道尔顿的聚乙二醇,其中平均分子量不大于标称数值的110%,并且不小于标称数值90%(优选地,不大于105%并且不小于95%)。聚乙二醇为式h
‑‑
[och2ch2]n‑‑
oh。n的平均值为至少3,并且一般为3至10,诸如3至6(虽然该平均不需要为整数)。聚乙二醇可从商业供应商以制药级广泛获得,并且以特定的标称分子量销售,这一般表明平均分子量不超过标称数值的105%并且不小于标称数值的95%。粘度和分子量测定方法公开于usp nf official compendium of standards(卷11180-1182[2007版])中。在一组实施方式中,聚乙二醇的分子量为200至400。在一些实施方式中,可优选地使用特定纯的乙二醇低聚物,诸如如下式的化合物
[0057]
h—[och2ch2]n—oh其中n为3或4。
[0058]
本发明还提供治疗患有选自微生物感染、病毒感染和癌症的疾病的受试者的方法,该方法包含对受试者给药有效量的根据所述工艺制备的生物活性试剂。
[0059]
本发明还可表示为丙烯醛和聚亚烷基二醇在制造用于治疗选自细菌感染、病毒感染和癌症的疾病的药剂中的用途,其中该用途包含所述工艺。
[0060]
通过所述工艺制备的共聚物特别地适用于给药以治疗选自癌症、病毒感染或细菌感染的肠胃外疾病。
[0061]
该共聚物典型地系统(全身)给药,例如通过口服给药,吸入,经皮递送或通过注射诸如到血流中、或肌肉内注射或通过静脉疗法,诸如通过注射或输注。一般认为,分子量不大于约1000、特别地小于约800道尔顿的分子具有穿过腹膜的合理自由通过。口服给药要求共聚物通过肠壁吸收并进入系统(全身)循环。在此实施方式中,特别优选的是口服给药的共聚物分子量不大于1000道尔顿,诸如分子量在250至800道尔顿、诸如300至800或350至800的范围中。我们已经发现,此分子量的共聚物在口服给药时被输送至系统(全身)循环中,以提供肠胃外病毒感染的治疗。对于具有在此范围中的较低分子量的共聚物而言,通过肠壁吸收的共聚物的比例一般较大。
[0062]
共聚物可作为气雾剂(气溶胶)、凝胶、局部泡沫或软膏施用或者浸渍到用于施用至皮肤或粘膜的敷料中以经皮或穿粘膜递送。共聚物可经由气雾剂等作为吸入剂施用。
[0063]
在进一步的实施方式中,共聚物通过经皮递送由可包括用于聚合物的渗透促进剂的组合物给药。贴片(patch)、微针(显微针)或类似装置可用于增强经皮递送。
[0064]
本发明的组合物还可包含佐剂(辅剂),例如防腐剂、润湿剂、乳化剂和分散剂。通过包括各种抗细菌剂和抗真菌剂例如对羟基苯甲酸酯、氯丁醇、苯酚、山梨酸等可确保防止微生物的作用。包括调节张度(tonicity)的试剂例如糖、氯化钠等也可为合乎需要的。可注射药物形式的延长(长期)吸收可通过包括延迟吸收的试剂例如单硬脂酸铝和明胶来实现。
[0065]
在另一优选的实施方式中,药物组合物呈适于皮下(s.c.)给药的形式。
[0066]
适于口服给药的药物剂型包括片剂(包衣的或未包衣的)、胶囊(硬壳或软壳)、囊片(小胶囊)、丸剂、锭剂、糖浆剂、溶液、散剂(粉末)、颗粒剂、酏剂和混悬液、舌下片剂、包药干糊片(糯米纸囊剂,wafer)或贴片例如口腔贴片。
[0067]
由于所述共聚物是可溶的且在1至14的整个ph范围内保持可溶的,其可以含水组合物配制。共聚物可以具有已知的药学上可接受的载体和赋形剂的组合物给药;然而,含水制剂提供显著的优点。取决于待治疗的特定病毒和给药方式,组合物可包括宽范围的浓度的共聚物。在一组实施方式中,含水药物组合物中的共聚物的浓度在组合物的0.01重量%至20重量%的范围内。因此,在一组优选实施方式中,共聚物作为水溶液给药。
[0068]
组合物可以片剂、囊片、糖浆剂或液体的形式口服给药,并且口服给药的剂量将取决于病毒的严重性和类型,但可在例如1mg-1000mg/千克体重/天,例如10mg-500mg/千克体重/天的范围内。
[0069]
在进一步的方面中,本发明提供用于治疗选自细菌感染、病毒感染和癌症的受试者中疾病的组合物,其中所述组合物是通过本文中描述的工艺制备的。
[0070]
共聚物和治疗方法的显著优点之一是,它们可用于针对来自宽范围病原体的感染,并且特别地可用于治疗可快速恶化从而呈现严重威胁(诸如菌血症或败血症或肺炎或脑膜炎(meningitis)或蜂窝组织炎(cellulitis))的细菌感染。这样的细菌感染的具体实例可选自由如下细菌组成的组:变形杆菌(proteus spp),沙雷氏菌(serratia spp),铜绿假单胞菌(pseudomonas aeruginosa),脑膜炎奈瑟球菌(neisseria meningitidis),大肠杆菌,克雷白杆菌肺炎(klebsiella pneumonia),金黄色葡萄球菌,凝固酶阴性葡萄球菌(staphylococcus spp),化脓性链球菌(streptococcus pyogenes),化脓性肺炎(streptococcus pneumonia)和肠球菌(enterococcus spp)。
[0071]
针对宽范围的病原体、并且特别是针对宽范围的细菌的活性允许该共聚物在严重或威胁生命的感染中用作治疗的第一线,例如在所述严重或威胁生命的感染中,感染的严重性可无法允许足够的时间来正确识别作为原因的(responsible)细菌。
[0072]
病毒感染可由一系列病毒导致,所述病毒诸如带衣的(coated)病毒(例如,带磷脂衣的病毒),包括疱疹、hiv、巨细胞病毒属(cytomegalovirus)和流感。优选地,通过本发明的方法治疗和/或控制的病毒感染可为:hsv-1、hsv-2、水痘带状疱疹病毒(varicella zoster virus)(呈水痘(chicken pox)或带状疱疹(shingles)的形式)、hcmv、ebv、疱疹6、疱疹7、疱疹8和sars-cov-2。
[0073]
在另一实施方式中,病毒为流感病毒,诸如流感a。
[0074]
在还另进一步的实施方式中,病毒为罗斯河病毒(ross river virus)。
[0075]
在进一步实施方式中,病毒为冠状病毒,包括作为严重的极性呼吸综合征的原因的冠状病毒。
[0076]
在一个实施方式中,病毒感染为sars-cov-2,一般亦称为covid-19。
[0077]
在一组优选的实施方式中,本发明的制备共聚物的工艺包含以下步骤:
[0078]
提供聚亚烷基二醇(优选地分子量在范围200至600道尔顿中的聚乙二醇)的弱碱性的(优选地ph不大于12.5;更优选地ph 8至12.5,诸如9.0至12.0)水溶液;
[0079]
将弱碱性的溶液剧烈混合,以夹带空气;将丙烯醛作为浓度不大于丙烯醛水溶液
(通常含有防腐剂)的50%w/w的水溶液(优选地在例如至少2分钟、更优选地至少5分钟的时间段内缓慢地)添加;
[0080]
在添加丙烯醛期间,维持溶液在不大于15℃、优选地不大于12℃(诸如不大于10℃、或不大于8℃)的温度,以形成共聚物;
[0081]
以及优选地,一旦丙烯醛单体已耗尽,就添加酸以提供小于9、并且优选地不大于8的ph。
[0082]
优选地,通过在如下反应容器中进行反应而将温度控制到不大于15c、优选地不大于12c,所述反应容器设置有间接热交换器,诸如具有围绕反应容器的至少一部分的冷却剂流体流(诸如水流)的夹套。
[0083]
所得共聚物的分子量通过聚亚烷基二醇的分子量控制,并且与其羟基浓度呈正比。(聚合可在环境温度下开始,随后来自聚合反应的放热通过以下中的一种或多种控制:反应混合物的体积,溶剂(诸如水)的量,反应物的添加速率(诸如向含水聚亚烷基二醇添加丙烯醛的速率),以及使用具有间接热交换的反应容器。
[0084]
在反应期间,优选地持续搅拌,并且仅作为必需的维持ph弱碱性(优选地ph不大于12.5,更优选地ph 9至12.5、9至12、或9至11)。添加更多碱并且使其浓度最小化,以使降解/副反应减低,并且使产物中形成羰基或羧基减少。
[0085]
最后,溶液的ph值可降低。在一组优选实施方式中,通过添加酸将ph调节至接近中性。丙烯醛的极度刺激性气味在共聚物产物中不再是明显的,其通常以至少99%的产率形成。
[0086]
得到的丙烯醛-共聚物典型地具有在250至1000道尔顿(例如300至1000道尔顿、400至1000道尔顿或400至800道尔顿)的范围内的分子量。共聚物没有将由聚丙烯醛的任何内容物(含量)预料到的混浊性。在共聚物的其制备中使用的丙烯醛:聚乙二醇的重量比优选地在1:4和1:40之间,更优选地在1:8和1:20之间。
[0087]
优选的碱是碱金属氢氧化物的水溶液;更优选地,碱金属氢氧化物为氢氧化钠。
[0088]
优选的酸是稀盐酸,尽管乙酸对于ph缓冲的目的是有用的。
[0089]
优选的是,将丙烯醛加入到聚亚烷基二醇的水溶液中花费约10分钟,并且反应至完成(完全)一般花费约40分钟,且优选地不超过90分钟。
[0090]
典型地,我们已经发现50分钟的反应时间适于获得向所述共聚物产物的基本上完全转化。
[0091]
丙烯醛优选地作为水溶液加入到含水聚亚烷基二醇,更优选地以在10-30重量%的范围内的丙烯醛单体的浓度,基于待加入聚亚烷基二醇水溶液中的丙烯醛水溶液的重量。
[0092]
得到的共聚物具有小于10%、更优选地小于5%,并且还更优选0%的反应性羰基含量(加上任何羧基含量)。
[0093]
丙烯醛溶液通常包含抑制剂氢醌,例如不超过0.5%并且典型地0.01-0.5%,以及更优选地0.1%重量/重量。
[0094]
对于本领域技术人员明晰的是,本文中的共聚物可被包括在多种组合物和物理形式中。特别地,组合物和体内药学使用方法将是明晰的,其利用共聚物的较慢的清除率。另外,明晰的是,可在药理学上利用分子量的变化以调节穿过膜、组织和器官的穿透率(速
率)、以及所得的在人体或动物体内的吸收或分布;在这种情况下,较低分子量的共聚物如例如300-800道尔顿比超过1000道尔顿分子量的共聚物更快地被吸收和分布。
[0095]
鉴于本文中的结果,也可想到的是添加蛋白质、特别地肉汤(broth)以提高共聚物的使用中的抗微生物活性。
[0096]
本文中的主题产品是水溶性的,并且可通过医学中已知的常用方法向人/动物给药(特别地通过口或注射),以及可能够以任何实践性的药物方式,单独地或以组合物的形式,在器官和组织内、或与人或动物接触地或在其体内血管系统中使用。当向人和动物给药所述共聚物时,它们可以其本身或作为包含例如与药学上可接受的载体组合的0.1%至99.5%(更优选地0.5%至90%)的活性成分的药物组合物给药。
[0097]
本领域技术人员还知晓,细菌感染可导致癌症,或是由于感染导致慢性炎症,或是由于感染释放诱导癌症的代谢产物(幽门螺杆菌(helicobacter pylori)导致癌症是熟知的实例)。因而,由于已发现本发明内描述的共聚物作为针对细菌、病毒和癌症的抗生素药品是实用的,因此本文描述的活性应为有利的并且可能是协同的。此外,由于病毒感染,特别是由流感病毒导致的病毒感染,也常与细菌感染相关联,再一次地,在一个药品中并行的抗病毒和抗生素性质有利地是协同的。
[0098]
现将参考以下实施例进一步描述本发明。将理解,实施例是以说明本发明的方式提供的,并且它们绝不限制本发明的范围。
[0099]
实施例
[0100]
实施例1和对比实施例——制备约500道尔顿mw的共聚物
[0101]
以下工艺在其中受计算机控制的反应温度范围为10℃(根据本发明的组合物实施例e1)至40℃(对比实施例4)的容器中进行,如表1中所示。
[0102]
反应在玻璃反应器中进行,该玻璃反应器设置有搅拌器和水流夹套热交换以及在反应期间的自动化计算机反馈温度控制。
[0103]
表1
[0104][0105]
向水(20g)和聚乙二醇(60g;mw 200)的溶液(已通过添加1m含水氢氧化钠使其ph 12)缓慢添加新鲜蒸馏的丙烯醛(5g;使用氢醌0.1%w/w抑制)在水(20g)中的溶液,历经10分钟;在该10分钟期间,被氧化的氢醌的黄色快速出现,随后消失。在该工艺期间,持续并且剧烈地搅拌组合物,以提供与空气充裕的接触。发生放热且快速的聚合。
[0106]
在另外的50分钟之后,通过添加1m含水盐酸将澄清溶液调整至ph7.5;产物是澄清的,几乎无色的(非常淡的黄色)溶液。本文中所由对样品进行的测试和结果都是在未经任何纯化的样品上进行的。
[0107]
产物的uv-可见、200-600nm光谱仅在200-300nm区域的远边界中具有显著的吸收。
[0108]
hplc表明,聚合产率为99-100%w/w,并且任何残留的丙烯醛单体均小于1ppm w/w;当测试最低至ph 1(并且最高至ph 14)时,该共聚物保持可溶。
[0109]
对于实施例1和对比实施例1至4中的每一者的共聚物,确定金黄色葡萄球菌和大肠杆菌的mic,并且与在所示的不同波长处检测的uv峰一起于表2中报道。
[0110]
表2
[0111][0112]
实施例2——治疗烧伤感染
[0113]
此实施例考察了实施例1和对比实施例3的组合物用模式化合物(moded compound)在治疗伤口感染中的功效(有效性,efficacy),其中阴性对照没有任何治疗,且阳性对照使用沙弗霉素。
[0114]
程序
[0115]
实验设计
[0116]
表3
[0117][0118]
实验程序
[0119]
制备接种物[第-1天]
[0120]
在诱导感染前一天或者在制造伤口的当天,将甘油贮存物金黄色葡萄球菌[atcc 43300]培养物解冻并接种到新鲜的酪蛋白大豆消化物(csd)肉汤中,并且在37℃下在摇动培育器(200rpm)中培育过夜(优选地,接种是在晚上完成的)。
[0121]
产生烧伤伤口
[0122]
将6-8周龄wistar雄性大鼠的背部皮肤刮净。通过吸入2-3%异氟醚麻醉剂而将动物麻醉。通过掐尾部检查麻醉深度。使用加热最高至80℃的熔融蜡在经刮净和消毒的表面上产生烧伤伤口。通过2cm
×
2cm塑料模具将蜡倒在动物刮净的背上。允许蜡留在皮肤上直至其凝固。以10mg/kg的剂量皮下给药酮洛芬(ketoprofen),以降低疼痛和应激(stress)。在有给养的情况下将动物单独圈养。
[0123]
诱导感染
[0124]
在受伤1天之后,使用100μl的~108cfu/ml金黄色葡萄球菌在皮肤伤口的位点处
使大鼠感染,并且用无菌棉纱布覆盖伤口位点。
[0125]
制剂
[0126]
共聚物的载剂以0.9%盐水灭菌。共聚物测试剂量配制为:组2——383mg的实施例1添加至20ml载剂(浓度=19.15mg/ml),以及组3——383mg的对比实施例3添加至20ml载剂(浓度=19.15mg/ml)。
[0127]
治疗
[0128]
治疗在感染之后一天开始。使用测试和参照化合物治疗动物连续3天,如实验设计中所示。
[0129]
通过移液管向伤口感染区域施加100μl体积的实施例1(浓度=19.15mg/ml)和实施例3(浓度=19.15mg/ml)两者,之后使用无菌弯尖(bent tip)在伤口表面上将溶液均匀地从中心向周围末端扩散,并且用无菌棉纱布覆盖。
[0130]
使用无菌棉拭子对伤口表面施加沙弗霉素(30mg b.i.d.)。
[0131]
观察
[0132]
体重:在整个研究期间,每日记录体重。
[0133]
临床征兆:在整个研究期间,每日监测动物的临床征兆。
[0134]
伤口收缩
[0135]
在第0天和第4天,通过在透明纸上追踪伤口边界,测量伤口面积的变化。随后将踪迹转移至1mm2作图纸(graph sheet)上,由此评估伤口表面积。随后,通过使用以下等式采用评估的表面积计算伤口收缩的百分数:
[0136][0137]
上皮化时间段
[0138]
上皮化时间段标记为结痂掉落、未留下原始伤口所需的伤口愈合之后的天数。
[0139]
伤口的细菌学评估
[0140]
在受伤之后的第1天和第4天使用棉拭子擦拭整个伤口。随后将拭子切下,并且置于含有1ml的无菌生理盐水的管中。在使用涡流混合器混合以将细菌细胞从拭子释放至盐水中之后,将100μl的未稀释的细胞悬浮液或其100倍稀释液铺在csd琼脂板上,并在37℃下在csd琼脂板上培育过夜(16h),用于细菌计数。
[0141]
数据分析
[0142]
估算各动物的体重、伤口收缩百分数和细菌载量(load)。使用软件graphpad prism(v 5.0)使用恰当的统计测试评估了组和对照平均值之间的显著差异。
[0143]
结果
[0144]
评估了在使用金黄色葡萄球菌[atcc 4330]感染的大鼠中的烧伤伤口感染模型中,e1和ce3组合物的功效。
[0145]
表4.治疗之后皮肤上的细菌载量
[0146][0147]
*显著地低于第1天(p《0.05,成对的t检验)
[0148]
汇总
[0149]
·
组合物e1测试剂量(100μl(19.15mg/ml),局部,u.i.d,3天)显示,与第1天相比,第4天的细菌载量显著降低,而在载剂对照中,不存在细菌载量的显著降低(p》0.05)。
[0150]
·
ce3测试剂量(100μl(19.15mg/ml),局部,u.i.d,3天)显示,与e1相比第4天的功效显著更低。
[0151]
·
沙弗霉素(30mg,局部,b.i.d,q=12hr,3天),与第1天相比,在第4天时未显示显著的功效,尽管平均载量更低。
[0152]
结果绘制于图1中所示的图中。
[0153]
表5.伤口收缩百分数
[0154][0155]
*显著不同于载剂对照(p《0.05,单因素anova)
[0156]
汇总:
[0157]
·
当与载剂对照相比时,组合物e1测试剂量(100μl(19.15mg/ml),局部,u.i.d,3天)、组合物ce3测试剂量(100μl(19.15mg/ml),局部,u.i.d,3天)和沙弗霉素(30mg,局部,b.i.d,q=12hr,3天)显示,与第1天相比,在第4天时伤口显著减小(p《0.05)。
[0158]
结果绘制于图2中。
[0159]
实施例3——治疗上行尿道感染
[0160]
本实施例比较了在上行尿道感染大鼠模型中e1和ce3组合物针对大肠杆菌的功效。
[0161]
实验程序
[0162]
颈静脉的导管插入术[2]
[0163]
在感染之前24小时,使用氯胺酮和赛拉嗪(70+10mg/kg)将6-8周龄的雄性wistar大鼠麻醉。通过掐尾部确认麻醉。使用wahl宠物修剪器将背侧和腹侧颈部表面(3-4cm)刮净,并且使用70%乙醇将手术位点消毒。以背卧方式放置大鼠,并且在腹侧颈部侧上制造1-2cm切口;将颈静脉解剖,并使其从周围组织脱离。在静脉下放置两处手术结扎(一处在头端(cranial end),并且另一处在尾端(caudal end))。头端被紧紧地打结,使用弹簧剪在尾端提供小切口,并且将填充有肝素化的盐水(100iu/ml)的导管插入到血管的管腔中(大致25-35mm)。将足够的血抽取到套管中,以确保妥当的放置和开放(patency)。
[0164]
制备接种物[第-1天]
[0165]
在感染前一天(第-1天),将大肠杆菌[atcc 25922]的甘油贮存物接种至无菌酪蛋白大豆消化物肉汤中,并且在37℃保持,用于培育。
[0166]
第0天(感染日)
[0167]
在感染日,检查过夜培养物,将其离心,并且将片状细胞再悬浮于无菌生理盐水中,并连续稀释,以获得~1x109cfu/ml并且用于感染。将接种物在无菌csd肉汤中连续稀释10倍,并且对于各株系,将0.05ml的六份稀释液铺在预先培育的csd琼脂板上,以确定接种物的活体计数(cfu/ml)。
[0168]
诱导感染[3,4,5]
[0169]
选择具有100%开放的动物用于研究,并且将动物分成不同的组,如实验设计中所具体说明的。将经分组的动物笼子携带至作业间(procedure room),靠近生物安全舱。所有感染均在生物安全舱中进行,其具有恰当的个人防护。通过氯胺酮和赛拉嗪(60+10mg/kg i.p.)混合剂(cocktail)的腹腔内注射将动物麻醉。一旦动物处于足够深层次的麻醉,如通过足底反射监测的,就将各大鼠的腹壁用电动剃刀刮净,并且用10%聚维定碘(povidine iodine)清洁皮肤。在1.5至2cm下腹壁切口之后,通过钝器解剖(blunt dissection)将腹壁肌肉分离。将膀胱分隔并暴露,移去膀胱内的尿液,并且将0.1ml的无菌盐水或细菌培养物大肠杆菌(~1x108cfu/动物)注射至膀胱中。在将膀胱重新放回其原始位置之后,使用缝合线将腹部肌肉聚拢(approximate)并将皮肤闭合。使用10%聚维定碘清洁手术位点。
[0170]
制剂
[0171]
用于组合物e1的载剂为灭菌的0.9%盐水。美罗培南(meropenem)在milliq水中制备。
[0172]
治疗
[0173]
在感染之后4小时,将组4、5和6使用组合物e1以静脉内(颈静脉)方式作为单一剂量在有意识的动物中以恒定的输注速率(2ml/kg/hr)历经24小时的期间给药,剂量体积为48ml/kg。组合物e1的剂量水平为50、500和4000mg/kg。组2以静脉内(颈静脉)方式如下给药灭菌的0.9%盐水:作为单一剂量,以恒定的输注速率(2ml/kg/hr),在有意识的动物中历经24小时的期间,剂量体积为48ml/kg。美罗培南(组3)作为30mg/kg的单一iv推注给药(剂量体积为5ml/kg)。治疗的总持续时间为24小时。
[0174]
终止
[0175]
在治疗开始之后24小时(感染之后28小时),通过过量的co2在恰当的暴露室内使各组动物安乐死,如实验设计中所具体描述的。组1动物在感染之后4小时处死。
[0176]
收取组织和匀化
[0177]
将经安乐死的动物浸入70%乙醇中,用于表面去污。以无菌方式(aseptically)将器官移去——膀胱在尿道附近切去,并且通过钝器解剖将肾脏移去,以避免流血。通过使用匀化器(omni tip(thb220手持))将膀胱和每个肾脏分别地在无菌盐水中匀化,并且通过散布板技术通过使用csd琼脂板确定活体计数。在37℃下培育18-24小时之后,确定膀胱和肾脏的cfu/毫升匀化物。
[0178]
数据分析
[0179]
在各组中,估算了log
10
cfu/克膀胱和log
10
cfu/克肾脏。通过如下分析组平均值之
间的显著差异:单因素anova分析,继以dunnett多重对比检验,使用在95%置信水平的graphpad prism。《0.05的p值认为是显著的。
[0180]
[0181]
肾脏:
[0182]
·
当与载剂对照相比时,美罗培南在30mg/kg的单一iv推注剂量之后在肾脏中显示出显著的杀细菌活性。
[0183]
·
组合物e1[4000mg/kg]显示出毒性——所有动物均在输注开始12小时后死亡。
[0184]
·
在50和500mg/kg下,组合物e1显示出剂量依赖性抗菌效果,其中当与载剂对照相比时500mg/kg显示出显著的活性(p《0.05)。
[0185]
结果绘制于图3中。
[0186][0187]
膀胱
[0188]
·
当与载剂对照相比时,美罗培南在30mg/kg的单一iv推注剂量之后在膀胱中显示出显著的杀细菌活性。
[0189]
·
当与载剂对照相比时,组合物e1在50和500mg/kg下在膀胱中显示出显著的剂量依赖性效果(p《0.05)。
[0190]
膀胱感染的治疗结果显示于图4中。
[0191]
实施例4——流感a病毒感染
[0192]
概述
[0193]
本研究的目的是确定组合物e1和组合物ce3在静脉内(iv)给药(在c57bl/6小鼠中每日两次,持续5天)之后针对流感a病毒(h1n1)株系:a/nws/33(vr-219
tm
)的功效。在整个研究期间,使用e1和ce3治疗的小鼠显示出剂量依赖性体重改进。在第4天时,当与载剂对照相比时,利巴韦林、e1(25mg/kg、50和100mg/kg)、和ce3(100mg/kg、500和1000mg/kg)中的动物显示出显著更高的体重(p《0.05)。
[0194]
当与载剂对照相比时,利巴韦林显著地降低了肺部中的病毒生长速率和病毒载量;当与载剂对照相比时,e1显示出病毒生长速率和病毒载量方面的显著的剂量依赖性减少。当与载剂对照相比时,ce3显示出病毒生长速率和病毒载量方面的显著的剂量依赖性减少。在第4天时,当与载剂对照相比时,利巴韦林e1(25mg/kg、50和100mg/kg)和ce3(100mg/kg、500和1000mg/kg)中的动物显示出显著更低的病毒滴度(p《0.05)。
[0195]
在提供的溶液中,组合物ce3和组合物e1的浓度为1000mg/ml。
[0196]
测试体系
[0197]
物种:小鼠
[0198]
株系、性别:c57bl/6、雌性
[0199]
年龄:6-8周
[0200]
组数:9
[0201]
动物数/组:12
[0202]
总动物数:108
[0203]
病毒:流感a病毒(h1n1)株系:a/nws/33(vr-219tm)
[0204]
表8.实验设计
10分钟,在新鲜的无菌管中收集上清液,并在4℃或在-80℃下储存。
[0217]
临床观察
[0218]
监测动物的一般临床征兆、发病率和死亡率。在研究结束时测量体重。
[0219]
数据分析
[0220]
在各组中,估算平均值
±
sd log
10
pfu/g肺。通过如下分析组平均值和对照之间的显著差异:单因素anova,继以dunnett多重对比检验,使用在95%置信水平的graphpad prism。《0.05的p值认为是显著的。
[0221]
结果
[0222]
在第六天pi,载剂对照组中三只动物中的两只发现死亡。在组e1中(25mg/kg每日两次(iv推注,q12h),5日),三只动物中的两只虚弱,且三只动物中的一只发现死亡。在整个研究期间,所有其它组中的动物都看起来正常。
[0223]
在整个研究期间动物的体重显示于表9中。
[0224][0225]
未感染的对照中的动物获得重量;在整个研究期间载剂对照和e1(25mg/kg)组中
的动物显著地失去重量;阳性对照中的动物显示没有显著的体重减少。e1(50mg/kg和100mg/kg)组中的动物在第2天显示出平均体重的降低,但是显示出随后的体重增加;ce3(100mg/kg)中的动物在整个研究期间显示出体重减少,而500mg/kg和1000mg/kg在第6天显示没有显著降低。在第6天无法进行体重的统计分析,因为载剂对照组中小鼠数量为1。取而代之的是,该统计分析是在第4天完成的。在第4天时,当与载剂对照相比时,利巴韦林、e1(25mg/kg、50和100mg/kg)和ce3(100mg/kg、500和1000mg/kg)中的动物显示出显著更高的体重(p《0.05)。
[0226]
小鼠肺中的病毒滴度显示于表10中。
[0227][0228]
当与载剂对照相比时,利巴韦林显著降低了肺部中的病毒生长速率和病毒载量;当与载剂对照相比时,e1显示出肺部中病毒生长速率和病毒载量显著的剂量依赖性减少。当与载剂对照相比时,ce3显示出肺部中病毒生长速率和病毒载量显著的剂量依赖性减少。
[0229]
在第6天无法进行病毒滴度的统计学分析,因为载剂对照组中小鼠数量为1。取而代之的是,统计学分析是在第4天完成的。在第4天时,当与载剂对照相比时,利巴韦林、e1(25mg/kg、50和100mg/kg)和ce3(100mg/kg、500和1000mg/kg)中的动物显示出显著更低的病毒滴度(p《0.05)。
[0230]
结论
[0231]
在使用流感a病毒(h1n1)株系:a/nws/33(vr-219
tm
)感染的小鼠流感模型中,使用载剂治疗的动物显著失去重量;利巴韦林治疗的小鼠显示没有显著的体重减少;在整个研究期间使用e1和ce3治疗的小鼠显示出剂量依赖性体重改进。在第4天,当与载剂对照相比时,利巴韦林、e1(25mg/kg、50和100mg/kg)、和ce3(100mg/kg、500和1000mg/kg)中的动物显示出显著更高的体重(p《0.05)。当与载剂对照相比时,利巴韦林显著降低了肺部中的病毒生长速率和病毒载量;当与载剂对照相比时,e1显示出肺部中的病毒生长速率和病毒载量显著剂量依赖性减少。当与载剂对照相比时,ce3显示出肺部中的病毒生长速率和病毒载量显著剂量依赖性减少。在第4天,当与载剂对照相比时,利巴韦林、e1(25mg/kg、50和100mg/kg)、和ce3(100mg/kg、500和1000mg/kg)中的动物显示出显著更低的病毒滴度(p《0.05)。
[0232]
实施例5——最大容忍剂量
[0233]
实施例5a——在小鼠中重复静脉内推注定量给药(给药,dosing)持续7天后的mtd。
[0234]
雌性balb/c小鼠使用载剂、ce3(5、50和500mg/kg q12h,7天)和e1(5、50和500mg/kg q12h,7天)以静脉内方式(推注)定量给药。在定量给药之后,观察动物的一般临床征兆。简言之,当以静脉内方式定量给药每日两次(q=12h)持续1星期时,发现小鼠中e1的mtd为500mg/kg。当以静脉内方式定量给药每日两次(q=12h)持续1星期时,发现小鼠中ce3的mtd为50mg/kg。
[0235]
材料和方法
[0236]
测试体系
[0237]
物种:小鼠
[0238]
株系、性别:balb/c、雌性
[0239]
年龄:6周
[0240]
组数:7
[0241]
动物数/组:5
[0242]
总动物数:35
[0243]
e1和ce3的组合物是作为浓度1000mg/ml的液体制剂提供的。
[0244]
表11.实验设计
[0245][0246]
实验程序
[0247]
使用不同剂量的载剂和测试物质对动物以口服方式进行定量给药,每日两次(q12 h)持续7天,如上文所示。观察动物,持续7天,并且就以下参数与载剂对照组比较:
[0248]
1.体重,在刚要定量给药之前以及直至定量给药后7天
[0249]
2.临床征兆(步态、姿势、发病率),在定量给药之前以及定量给药之后即刻直至7天。
[0250]
3.观察动物的死亡率。
[0251]
制剂
[0252]
使用0.9%生理盐水作为e1和ce3的载剂。
[0253]
ce3组合物
[0254]
5mg/kg:将2.0μl的制剂(1000mg/ml)稀释成2ml,其中添加1.998ml的0.9%生理盐水。
[0255]
50mg/kg:将20μl的制剂(1000mg/ml)稀释成2ml,使用1.980ml的0.9%生理盐水。
[0256]
500mg/kg:200μl的制剂(1000mg/ml)稀释成2ml,使用1.800ml的0.9%生理盐水。
[0257]
每天e1组合物
[0258]
5mg/kg:2.0μl的制剂(1000mg/ml)稀释成2ml,其中添加1.998ml的0.9%生理盐水。
[0259]
50mg/kg:20μl的制剂(1000mg/ml)稀释成2ml,使用1.980ml的0.9%生理盐水。
[0260]
500mg/kg:200μl的制剂(1000mg/ml)稀释成2ml,使用1.800ml的0.9%生理盐水。
[0261]
在每次定量给药之前新鲜地制备制剂。
[0262]
定量给药
[0263]
使用不同剂量的ce3和e1对动物以静脉内方式(推注)进行定量给药,每日两次(q12h),持续7天,如上文所示。剂量体积为5ml/kg。
[0264]
结果
[0265]
动物的临床征兆,总体病理学(gross pathology)和体重显示于表12和13中。
[0266]
[0267]
[0268][0269]
结论
[0270]
使用低剂量ce3(5和50mg/kg,i.v.,q12 h天,持续7天)治疗的小鼠在定量给药之
后看起来正常,并且可与载剂组相当。在使用ce3高剂量(500mg/kg,i.v.,q12 h天,持续7天)定量给药的小鼠中,五只小鼠中的三只在第4次定量给药之后发现死亡。
[0271]
在e1治疗的组(即,载剂,低、中等和高剂量组)中的动物在整个研究期间都看起来正常。
[0272]
在ce3和e1治疗小鼠的组(即,载剂,低、中等和高剂量组)两者中,平均体重均增加,ce3高剂量组除外。
[0273]
简言之,当以静脉内方式定量给药,每日两次(q=12h)持续1星期时,小鼠中e1的mtd发现为500mg/kg。当以静脉内方式定量给药,每日两次(q—12h)持续1星期时,小鼠中ce3的mtd发现为50mg/kg。
[0274]
实施例5b——在大鼠中重复口服定量给药持续7天后,确定实施例ce3和e1的组合物的最大容忍剂量mtd
[0275]
汇总
[0276]
雄性sprague dawley大鼠使用载剂、ce3组合物(5、50和500mg/kg q12h)和e1组合物(5、50和500mg/kg q12h)以口服方式定量给药,持续7天。定量给药后,观察动物的一般临床征兆和体重。在大鼠中,当以口服方式给药,每日两次重复持续7天时,发现ce3和e1的最大容忍剂量mtd为至少500mg/kg。
[0277]
测试
[0278]
ce3和e1的组合物作为玻璃瓶中浓度1000mg/ml的溶液供应。
[0279]
测试对象
[0280]
物种:大鼠
[0281]
株系:sprague dawley
[0282]
年龄:6
–
8周
[0283]
性别:雄性
[0284]
实验程序
[0285]
动物分成不同组,如实验设计中所示,并且随意提供饲料和水。使用不同剂量的测试物质对大鼠组以口服方式定量给药,每日两次(q12h),持续7天,如上文所示。
[0286]
结果
[0287]
在所有剂量下,使用载剂、ce3和e1定量给药的动物在整个研究期间看起来正常(表1)。在所有组中,动物的平均体重增加,并且所有组中都没有观察到总体病理学的明显异常。
[0288]
结论
[0289]
每日重复,持续7天,发现在大鼠中为至少500mg/kg。当ce1和e1组合物以口服方式两次时的mtd
[0290]
实施例6——sars-cov-2
[0291]
在syrian金仓鼠(一种被广为接收的感染模型)中,e1和ce2的组合物在体内展现了针对sars-cov-2的剂量依赖性活性。
[0292]
这两种化合物的鼻内给药均支持了针对sars-cov-2的多种潜在给药模式。
[0293]
方法
[0294]
本研究由五个八只仓鼠组组成,各组接受不同的治疗——盐水鼻洗剂的安慰剂对
照,低剂量的ce3(200mg/kg),高剂量的ce3(400mg/kg),低剂量的e1(100mg/kg),和高剂量的e1(200mg/kg)。在第0天,所有动物均使用sars cov-2感染,在第1-5天实施治疗,每日两次,并且在第2、4和6天经由qpcr直接测量病毒滴度。在此模型中,在第2和4天之间,病毒滴度典型地达到峰值。
[0295]
与安慰剂组相比,在ce3和e1两者中的结果证明了covid-19病毒载量的阳性降低。结果显示于附图的图5中,其为显示使用sars-cov-2感染并且使用组合物e1和ce3治疗的仓鼠中鼻洗剂滴度(log
10
染色体组/μl)的图。在图中,对于每一天而言,图从左至右涉及安慰剂、ce3(200mg/kg)、ce3(400mg/kg)、e1(100mg/kg)和e1(200mg/kg)。第4天组内的平均值对数降低,其中低e1剂量实现了约1.5对数的对数降低,并且高剂量的ce3实现了1.25对数的对数降低。在第6天时,高剂量e1组中感染covid-19的五只仓鼠中的两只表现不良的临床症状,并从研究中排除。公司认为研究特别异常,因为在体内在相当高的血管内输注剂量下e1通常是被良好容忍的。在研究开始和结束时,所有组中的仓鼠重量保持大致相等。
[0296]
此仓鼠研究证明了e1经鼻给药(nasal administration)的潜力,尤其是当用于针对病毒时。
[0297]
e1证明了在低剂量下针对sars-cov-2的活性较高。因而,与ce3相比,e1组合物呈现出优越的体内抗病毒功效,这与对于在小于15℃的温度下制备的共聚物所记录的较低mic一致。
[0298]
实施例7——sars-cov-2——人类气道上皮(hae)细胞
[0299]
部分a
[0300]
covid类器官方案:包含人类气道上皮(hae)细胞的类器官通过使用sars-cov-2病毒接种而感染,并且在37℃下使用不同浓度的ce3(h:31ppm、m:10ppm、l:3ppm)和e1(h:82ppm、m:41ppm、l:8ppm)培育,并且病毒载量通过在时间点评估的pfu(病毒的斑块形成单位)数测量。对照为聚乙二醇(peg)200。
[0301]
数据表明,与对照组相比,通过ce1和e1实现了自sars-cov-2(covid-19)病毒基线的浓度依赖性降低。sars-cov-2病毒是全球covid-19流行病的原因。使用的浓度与关于ce3静脉内输注程序的共聚物的那套临床前数据相比低得多。
[0302]
在两种共聚物组合物浓度不同的情况下,确定病毒感染中的浓度依赖性降低,并且显示于附图的图6中。
[0303]
数据表明与ce3相比,组合物e1针对sars-cov-2的活性更高,特别是在低剂量下。
[0304]
部分b
[0305]
在单独的研究中,ce3和e1表明卓越的毒性廓线(profile),其中在所测试的浓度下对vero(猴)细胞的影响小于0.25%。
[0306]
vero细胞方案中细胞毒性测试:在vero细胞发光检测中,评估了一系列浓度的ce3(153ppm、76ppm、38ppm、19ppm、13ppm、6ppm、3ppm、2ppm、1ppm)and e1(82ppm、55ppm、41ppm、33ppm、27ppm、16ppm、12ppm、8ppm、4ppm)的细胞毒性,并且在1小时、24小时和72小时培育的时间点处测量细胞存活(活力),使用的对照为未处理的健康vero细胞。在测量的所有时间点和浓度下,两种化合物均证明了最小的细胞毒性效应,其中大于99%的受试细胞保持其存活。
[0307]
细胞毒性测试的结果显示于图7(对于e1)和图8(对于ce3)中。对于各浓度,(从左
至右)对于1小时、24小时、和72小时报道了最大细胞毒性百分数。
[0308]
实施例8——小鼠中e1和ce3针对淋病奈瑟菌(neisseria gonorrhoea)(atcc700825)的功效
[0309]
汇总
[0310]
本研究的目的是在小鼠阴道感染模型中,评估实施例e1和对比实施例ce3的组合物针对淋病奈瑟菌(atcc700825)的功效。当与载剂对照相比时,美罗培南显示出在50mg/kg的iv推注定量给药、持续7天之后在阴道载量中显著的杀细菌活性(p《0.05)。e1显示剂量依赖性抗细菌效果。当与载剂对照相比时,在25和50mg/kg(iv推注,持续7天)剂量下的e1显示出在细菌载量方面平均值剂量依赖性降低,但是它们并不特别显著;在100mg/kg(iv推注,持续7天)下的e1显示出显著的抗细菌效果(p《0.05)。
[0311]
当与载剂对照七天pi相比时,ce3在100、500和1000mg/kg(iv推注,持续7天)下显示出在阴道载量方面显著的剂量依赖性抗细菌效果(p《0.05)。
[0312]
测试项目
[0313]
ce3和e1呈1000mg/ml的水溶液。
[0314][0315][0316]
淋病奈瑟菌接种
[0317]
处在动情期周期的动情阶段的动物以5mg、21天受控释放的雌二醇药片以皮下方式植入,并且在整个感染期间使用链霉素(streptomycin)和甲氧苄啶(trimethoprim)处理,以增加其对淋病奈瑟菌的易感性。在植入药片两天之后,小鼠以阴道内方式被接种淋病奈瑟菌(~2
×
106cfu/动物)。
[0318]
制剂
[0319]
ce3、e1和美罗培南的载剂为灭菌的0.9%盐水。使用载剂将测试化合物稀释成所需的浓度。
[0320]
治疗
[0321]
感染之后两天,治疗动物,持续连续7天,如实验设计中所示。
[0322]
表14——使用测试化合物治疗的经感染的小鼠中的临床征兆。
[0323][0324]
n:看起来正常
[0325]
结果描绘于图9中,其显示了在小鼠中使用参照和测试项目治疗之后阴道拭子中的细菌载量。*p《0.05,显著不同于载剂对照。
[0326]
结论
[0327]
在使用淋病奈瑟菌(atcc700825)感染的小鼠阴道感染模型中,在50mg/kg的iv推注定量给药、持续7天之后,当与载剂对照相比时,美罗培南在阴道载量方面显示出显著的杀细菌活性(p《0.05)。
[0328]
e1显示出剂量依赖性抗细菌效果。当与载剂对照相比时,在25和50mg/kg(iv推注,持续7天)剂量下的e1显示出在细菌载量方面平均值剂量依赖性减少;在100mg/kg(iv推注,持续7天)下的e1显示出显著的抗细菌效果(p《0.05)。
[0329]
当与载剂对照七天pi相比时,在100、500和1000mg/kg(iv推注,持续7天)下的ce3显示出阴道载量方面显著的剂量依赖性抗细菌效果(p《0.05)。
[0330]
因而,与ce3相比,e1组合物呈现出优越的体内功效,这与对于在小于15℃的温度下制备的共聚物记录的mic较低一致。
[0331]
实施例9——在5℃和10℃下制备的本发明的组合物的mic确定
[0332]
使用实施例1的程序,在5℃(实施例2
–
e2)和10℃(实施例1,e1)下制备本发明的组合物。
[0333]
对实施例1和实施例2中的每一者的共聚物确定金黄色葡萄球菌和大肠杆菌的mic,于表15中报道。
[0334]
表15
[0335]
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1