制备雄激素受体拮抗剂及其中间体的方法与流程
1.本发明涉及制备2-氯-4-(1-(四氢-2h-吡喃-2-基)-1h-吡唑-5-基)苄腈(iii)的改进方法,其可用作制备甲酰胺结构的雄激素受体拮抗剂如n-((s)-1-(3-(3-氯-4-氰基苯基)-1h-吡唑-1-基)-丙-2-基)-5-(1-羟基乙基)-1h-吡唑-3-甲酰胺(1a)的中间体。
背景技术:
2.式(1a)的化合物n-((s)-1-(3-(3-氯-4-氰基苯基)-1h-吡唑-1-基)-丙-2-基)-5-(1-羟基乙基)-1h-吡唑-3-甲酰胺及其衍生物公开于wo 2011/051540中。式(1a)化合物及其衍生物是可用于治疗癌症、特别是前列腺癌和需要ar拮抗作用的其它疾病的有效的雄激素受体(ar)拮抗剂。
[0003][0004]
wo 2011/051540公开了经由式(iii)、(iv)和(v)的中间体制备式(1a)化合物的方法,如流程i所示:
[0005][0006]
流程i
[0007]
通过将1-(四氢-2h-吡喃-2-基)-1h-吡唑-5-硼酸频哪醇酯(i)与4-溴-2-氯苄腈(ii)在suzuki反应中反应来制备式(iii)化合物或2-氯-4-(1-(四氢-2h-吡喃-2-基)-1h-吡唑-5-基)苄腈。suzuki反应在均相(可溶性)双(三苯基膦)氯化钯(ii)催化剂和碳酸钠碱存在下在thf-水溶剂中进行。反应完成后,将溶剂蒸馏至几乎干燥,并加入水以沉淀式(iii)化合物。
[0008]
wo 2012/143599中公开了制备式(iii)化合物的类似方法。suzuki反应在均相双(三苯基膦)氯化钯(ii)催化剂、碳酸钠碱和相转移催化剂(tbab)的存在下在thf-甲苯-水溶剂中进行。式(iii)化合物的分离通过加入水并将分离的有机相蒸馏至接近干燥,随后加入乙醇并过滤结晶产物来进行。
[0009]
最后,wo 2016/162604描述了制备式(iii)化合物的方法,其中suzuki反应在均相pd(oac)2催化剂、碳酸钾碱和三苯基膦存在下在乙腈-水溶剂中进行。通过从反应混合物中除去水相、加入氨水(25%)并冷却反应混合物、随后加入水并分离结晶产物来分离出式(iii)化合物。
[0010]
上述方法的缺点是昂贵的可溶性钯催化剂在反应后被废弃,这占了生产成本的相当一部分,并且在分离出的产物中残留有痕量的钯催化剂。
[0011]
因此,需要一种更实用和经济的适于大规模制备ar拮抗剂中间体例如式(iii)化合物的方法。
技术实现要素:
[0012]
现已发现,式(iii)化合物可通过多相催化剂大规模制备,导致最终产物的高产率、高纯度和短的反应时间。由于多相催化剂被固定或负载在固体载体上,因此它易于回收和再循环,从而显著降低该方法的生产成本。在最终产物中发现的催化剂残留物的水平也显著降低。
[0013]
因此,本发明提供了制备式(iii)的2-氯-4-(1-(四氢-2h-吡喃-2-基)-1h-吡唑-5-基)苄腈的方法
[0014][0015]
该方法包括将式(ia)或(ib)的化合物
[0016][0017]
其中r1和r2是氢,或者r1和r2一起形成直链或支链的c
2-6
烷基链或-c(o)-ch
2-n(ch3)-ch
2-c(o)-链,
[0018]
与式(ii)的4-溴-2-氯苄腈
[0019][0020]
在高温下在多相钯催化剂、溶剂和碱的存在下进行反应。
[0021]
另一方面,本发明提供了一种制备式(v)的2-氯-4-(1h-吡唑-3-基)苄腈的方法
[0022][0023]
该方法包括以下步骤:
[0024]
(a)将式(ia)或(ib)化合物
[0025]
[0026]
其中r1和r2是氢,或者r1和r2一起形成直链或支链的c
2-6
烷基链或-c(o)-ch
2-n(ch3)-ch
2-c(o)-链,
[0027]
与式(ii)的4-溴-2-氯苄腈
[0028][0029]
在高温下在多相钯催化剂、溶剂和碱的存在下反应,得到式(iii)化合物
[0030][0031]
(b)将式(iii)化合物用hcl处理;
[0032]
(c)加入碱以得到式(v)化合物。
[0033]
在另一方面,本发明提供了制备式(1a)化合物的方法
[0034][0035]
该方法包括以下步骤
[0036]
(a)将式(ia)或(ib)化合物
[0037][0038]
其中r1和r2是氢,或者r1和r2一起形成直链或支链的c
2-6
烷基链或-c(o)-ch
2-n(ch3)-ch
2-c(o)-链,
[0039]
与式(ii)的4-溴-2-氯苄腈
[0040][0041]
在高温下在多相钯催化剂、溶剂和碱的存在下反应,得到式(iii)化合物
[0042][0043]
(b)将式(iii)化合物用hcl处理;
[0044]
(c)加入碱以得到式(v)化合物
[0045][0046]
(d)将式(v)化合物与式(vi)化合物反应
[0047][0048]
生成式(vii)化合物;
[0049][0050]
(e)将式(vii)化合物与式(viii)化合物反应
[0051][0052]
生成式(ix)化合物;然后
[0053][0054]
(f)还原式(ix)化合物以制得式(1a)化合物。
具体实施方式
[0055]
本文所用的术语“多相钯催化剂”是指固定或负载在固体载体上从而在反应完成后可容易地例如通过过滤从反应介质中除去的钯催化剂。
[0056]
本文所用的术语“钯的摩尔%”是指反应步骤中所用的钯量(以摩尔计)相对于起始化合物的量(以摩尔计)的百分比。例如,如果在反应中每1摩尔溴-2-氯苄腈使用0.005摩尔的钯,则所使用的钯的摩尔%为(0.005/1)*100摩尔%=0.5摩尔%。
[0057]
互变异构:由于吡唑环的氢原子可以存在于1-和2-位之间的互变异构平衡中,本领域技术人员认识到,本文所公开的在吡唑环中包含氢原子的化学式和化学名称包括所讨论的化合物的互变异构体。例如,化学名为“2-氯-4-(1h-吡唑-3-基)苄腈”和相应的式(v)包括该化合物的互变异构体,即“2-氯-4-(1h-吡唑-5-基)苄腈”。
[0058]
根据本发明,式(iii)的2-氯-4-(1-(四氢-2h-吡喃-2-基)-1h-吡唑-5-基)苄腈
[0059][0060]
通过将式(ia)或(ib)化合物
[0061][0062]
其中r1和r2是氢,或者r1和r2一起形成直链或支链的c
2-6
烷基链或-c(o)-ch
2-n(ch3)-ch
2-c(o)-链,
[0063]
与式(ii)的4-溴-2-氯苄腈
[0064][0065]
在高温下在多相钯催化剂、溶剂和碱的存在下进行反应来制备。
[0066]
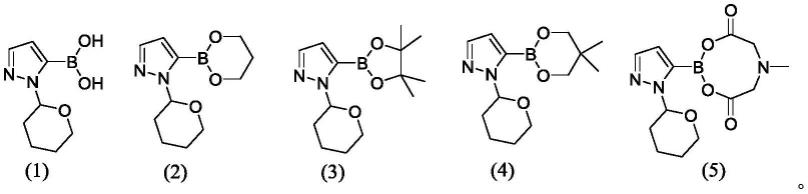
根据本发明的一个优选实施方案,式(ia)化合物选自下列化合物:
[0067][0068]
根据本发明的特别优选的实施方案,将式(ii)的4-溴-2-氯苄腈与式(ia)化合物即1-(四氢-2h-吡喃-2-基)-5-(4,4,5,5-四甲基-1,3,2-二氧杂硼杂环戊烷-2-基)-1h-吡唑(3)进行反应。
[0069]
反应中使用的多相钯催化剂是固定或负载在固体载体上的钯催化剂。多相钯催化剂的实例包括钯/碳、钯/硫酸钡、钯/金属氧化物(例如氧化铝)、钯/硅或钯/沸石。多相钯催化剂是可商购的,例如以商标购买自evonik industries ag。实例包括p1064(5%钯/活性炭)、p1070(10%钯/活性炭)、p1090(5%钯/活性炭)、p1092(5%钯/活性炭)、p1093(5%钯/活性炭)和p1095(5%钯/活性炭),它们可以以湿的自由流动粉末的形式获得。在本发明的方法中,相对于式(ii)化合物的量所用的钯的量通常为约0.2至约1mol%,优选约0.4至约0.8mol%,例如0.5mol%。反应优选在不存在钯配体如三苯基膦的情况下进行,因为发现当使用多相钯催化时,这些配体会干扰反应。
[0070]
反应在适当的溶剂中进行。尽管可以使用任何适当的溶剂,但溶剂优选仅包含二
甲亚砜(dmso),或更优选包含二甲亚砜与水的混合物。合适地,水与dmso的体积比为约0:100至约50:50,优选约1:99至约35:65,更优选约5:95至约20:80,例如10:90。
[0071]
特别适合进行反应的碱是有机碱,包括三烷基胺,例如二异丙基乙胺(dipea)、三甲胺(tea)或三丁胺(tba)。优选三烷基胺,特别是二异丙基乙胺(dipea),其适当的用量相对于化合物(ii)为1-2摩尔当量,例如1.3-1.6摩尔当量。
[0072]
反应优选在相转移催化剂如季铵盐存在下进行。特别优选四丁基溴化铵、四丁基氯化铵。
[0073]
根据本发明的一个特别优选的实施方案,反应在dmso-水溶剂中,在碱和相转移催化剂的存在下进行,所述碱是二异丙基乙胺(dipea),所述相转移催化剂是四丁基溴化铵或四丁基氯化铵。
[0074]
式(ia)、(ib)和(ii)化合物可商购获得,或者它们可按照本领域已知的方法制备。
[0075]
为了进行suzuki反应,可以首先在氮气氛下搅拌4-溴-2-氯苄腈(ii)、式(ia)或(ib)化合物(例如1-(四氢-2h-吡喃-2-基)-5-(4,4,5,5-四甲基-1,3,2-二氧杂硼杂环戊烷-2-基)-1h-吡唑(3))、溶剂、碱和相转移催化剂的混合物。反应适宜在氮气流下进行。加入催化剂,并将混合物加热至约60℃至约100℃,优选约70℃至约80℃,例如约72℃至约78℃的温度。搅拌混合物直到反应完成,例如约1至约5小时,通常约2至约4小时,然后,将混合物适当地冷却至约50-70℃,并例如通过在氮气压力下过滤除去多相钯催化剂。为了便于从反应混合物中除去多相钯催化剂,例如碳载钯,可以在过滤前向反应混合物中加入乙醇。发现碳载钯颗粒可在dmso中形成非常细的分散体,妨碍通过过滤从反应混合物中完全除去催化剂颗粒。发现乙醇的加入导致细催化剂颗粒聚集成更大的颗粒,其更容易通过过滤除去。过滤前的dmso:乙醇的适当比例为约10:2至约10:10,更典型地为约10:3至约10:5,例如约10:4。
[0076]
然后将滤液的温度适当地调节至约30-50℃,通过向冷却的混合物中缓慢地加入水来进行化合物(iii)的沉淀。加入的水量合适地为进行反应的溶剂的约60-120体积%,例如约65-80体积%。然后将所得悬浮液进一步冷却至约15-25℃,并搅拌完成化合物(iii)沉淀所需的时间,例如约3-12小时。沉淀的产物可以例如通过过滤分离,然后用水洗涤并干燥,例如在约40-60℃下减压干燥。该方法通常得到hplc纯度为99.5%或更高,更通常为约99.8%的化合物(iii)。
[0077]
式(iii)化合物向式(v)化合物的转化可以使用本领域已知的方法进行。例如,将溶于甲醇的式(iii)化合物用少量30% hcl(水溶液)在适当的低温如0-15℃下处理。在该温度下将混合物搅拌一段使四氢吡喃环发生脱离所需的时间,例如2小时。然后在上述温度下将碱例如氨水(25%)加入到混合物中。然后,在例如10-20℃下逐渐加入水,然后搅拌例如6-24小时。将式(v)化合物通过冷却该混合物例如冷却到约0-5℃进行沉淀,然后在该温度下搅拌足以完成沉淀的时间,例如合适地为约3至约5小时。例如通过过滤分离沉淀的产物。
[0078]
式(1a)化合物可以由式(v)化合物制备,例如使用wo 2011/051540和wo 2012/143599中所述的方法。例如,根据一个实施方案,制备式(1a)化合物的方法包括以下步骤:
[0079]
(d)将式(v)化合物
[0080][0081]
与式(vi)化合物反应
[0082][0083]
以生成式(vii)的化合物;
[0084][0085]
(e)将式(vii)化合物与式(viii)化合物反应
[0086][0087]
以生成式(ix)的化合物;然后
[0088][0089]
(f)将式(ix)化合物还原以制备式(1a)化合物。
[0090]
步骤(d)的反应可以例如使用mitsunobu反应条件进行,例如在室温下在三苯基膦和diad(偶氮二甲酸二异丙酯)的存在下在适当的溶剂例如thf或etoac中进行,然后通过用hcl处理进行boc-脱保护,最后用碱例如naoh处理。
[0091]
反应步骤(e)可以在室温下、在适当的活化剂和偶联剂体系例如dipea(n,n-二异丙基乙胺)、edci(1-乙基-3-(3-二甲基氨基丙基)碳二亚胺)和无水hobt(1-羟基-苯并三唑)的组合的存在下、在适当的溶剂(例如dcm)中进行。作为hobt的替代物,可以使用hbtu(六氟磷酸o-(苯并三唑-1-基)-n,n,n',n'-四甲基脲鎓)。另外,dipea和t3p(1-丙烷膦酸环酐)的组合可用作活化剂和偶联剂体系。
[0092]
反应步骤(f)可以在室温下进行,即在适当的溶剂如乙醇中用还原剂如硼氢化钠处理式(ix)化合物,然后用hcl水溶液处理该混合物。
[0093]
本发明通过以下非限制性实施例进一步说明。
[0094]
实施例1.在dmso/水溶剂中使用钯碳制备2-氯-4-(1-(四氢-2h-吡喃-2-基)-1h-吡唑-5-基)苄腈(iii)
[0095]
在氮气下向烧瓶中加入4-溴-2-氯苄腈(ii)(20g,1摩尔当量)、1-(四氢-2h-吡喃-2-基)-5-(4,4,5,5-四甲基-1,3,2-二氧杂硼杂环戊烷-2-基)-1h-吡唑(3)(28.4g,1.05摩
尔当量)、四丁基溴化铵(1.49g,0.05摩尔当量)、二甲亚砜(87.5ml)、水(12.5ml)和二异丙基乙胺(24.1ml,1.5摩尔当量)。将混合物通过使用真空抽空、随后再引入氮气进行脱气,同时剧烈搅拌。重复该过程三次。加入催化剂(5%钯碳,水湿的,1.0g干重,0.005摩尔当量),并将混合物在2小时内加热至75℃。将混合物搅拌直至反应完成(2-3小时),然后将混合物冷却至65℃。加入硅藻土(2g)和乙醇(40ml),并将混合物进一步搅拌约1小时。在氮气压力下通过过滤除去催化剂,将滤饼用二甲基亚砜(10ml)洗涤。将滤液的温度调节到45℃。在约30分钟内缓慢加入水(67ml)。将所得悬浮液冷却至20℃并将产物通过过滤收集。用水(40ml)洗涤滤饼,然后用冷乙醇(20ml)洗涤。在50℃真空干燥产物,得到24.5g(92%)纯度为99.8a%的标题化合物(iii)。
[0096]
实施例2.在dmso/水溶剂中使用钯/氧化铝制备2-氯-4-(1-(四氢-2h-吡喃-2-基)-1h-吡唑-5-基)苄腈(iii)
[0097]
在氮气下向烧瓶中加入4-溴-2-氯苄腈(ii)(5g,1摩尔当量)、1-(四氢-2h-吡喃-2-基)-5-(4,4,5,5-四甲基-1,3,2-二氧杂硼杂环戊烷-2-基)-1h-吡唑(3)(7.1g,1.05摩尔当量)、四丁基溴化铵(0.37g,0.05摩尔当量)、二甲亚砜(42.5ml)、水(7.5ml)和二异丙基乙胺(6.1ml,1.5摩尔当量)。将混合物通过使用真空抽空、随后再引入氮气进行脱气,同时剧烈搅拌。重复该过程三次。加入催化剂(5%钯/氧化铝,0.37g干重,0.0075摩尔当量),然后将混合物在30分钟内加热至75℃。将混合物搅拌直至反应完成(2-3小时),然后将混合物冷却至50℃,通过在氮气压力下过滤除去催化剂,将滤饼用二甲基亚砜(5ml)洗涤。将滤液的温度调节至35℃。在约30分钟内缓慢加入水(40ml)。将所得悬浮液冷却至20℃并通过过滤收集产物。用水(25ml)洗涤滤饼。在50℃真空干燥产物,得到6.4g(95%)纯度为99.8a%的标题化合物(iii)。
[0098]
实施例3.在dmso/水溶剂中使用钯碳制备2-氯-4-(1-(四氢-2h-吡喃-2-基)-1h-吡唑-5-基)苄腈(iii)
[0099]
在氮气下向烧瓶中加入4-溴-2-氯苄腈(ii)(5g,1摩尔当量)、1-(四氢-2h-吡喃-2-基)-5-(4,4,5,5-四甲基-1,3,2-二氧杂硼杂环戊烷-2-基)-1h-吡唑(3)(7.1g,1.05摩尔当量)、四丁基氯化铵(0.32g,0.05摩尔当量)、二甲亚砜(42.5ml)、水(7.5ml)和二异丙基乙胺(6.1ml,1.5摩尔当量)。将混合物通过使用真空抽空、随后再引入氮气进行脱气,同时剧烈搅拌。重复该过程三次。加入催化剂(5%钯碳,水湿的,0.25g干重,0.005摩尔当量),然后将混合物在30分钟内加热至75℃。搅拌混合物直至反应完成(2-3小时),然后将混合物冷却至50℃,通过在氮气压力下过滤除去催化剂,将滤饼用二甲基亚砜(5ml)洗涤。将滤液的温度调节至35℃。在约30分钟内缓慢加入水(40ml)。将所得悬浮液冷却至20℃并通过过滤收集产物。用水(25ml)洗涤滤饼。在50℃真空干燥产物,得到6.2g(93%)纯度为99.8a%的标题化合物(iii)。
[0100]
实施例4.在乙腈/水溶剂中使用钯碳制备2-氯-4-(1-(四氢-2h-吡喃-2-基)-1h-吡唑-5-基)苄腈(iii)
[0101]
在氮气下向烧瓶中加入4-溴-2-氯苄腈(ii)(5g,1摩尔当量)、1-(四氢-2h-吡喃-2-基)-5-(4,4,5,5-四甲基-1,3,2-二氧杂硼杂环戊烷-2-基)-1h-吡唑(3)(7.1g,1.05摩尔当量)、乙腈(27ml)、水(18ml)和碳酸钾(4.5g,1.4摩尔当量)。将混合物通过使用真空抽空、随后再引入氮气进行脱气,同时剧烈搅拌。重复该过程三次。加入催化剂(钯碳,1.0g干重,
0.02摩尔当量)和三苯基膦(0.49g,0.08当量)并将混合物加热至接近回流(约74℃)。将混合物搅拌2小时。此时分析表明4-溴-2-氯苄腈的转化率为14.4%,同时1-(四氢-2h-吡喃-2-基)-5-(4,4,5,5-四甲基-1,3,2-二氧杂硼杂环戊烷-2-基)-1h-吡唑(3)完全消耗,表明起始化合物(3)明显分解。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1