一种(+)-生物素的全连续流制备方法
1.本发明属于精细化工技术领域,具体涉及(+)-生物素的全连续流制备方法。
背景技术:
2.(+)-生物素(又称维生素h或辅酶r)的结构如式(1)所示
[0003][0004]
化学名称为(3as,4s,6ar)-六氢-2-氧代-1h-噻吩并[3,4-d]咪唑-4-正戊酸,是一 种水溶性b族维生素,主要参与机体碳水化合物、脂类、蛋白质和核酸等的代谢。人体缺 乏症状表现为皮炎、恶心、呕吐、情绪抑郁、体重减轻等;动物缺乏症状表现为皮屑、溃 疡、产仔率低、胚胎死亡率高,严重的直接引起动物死亡。补充适量(+)-生物素可以使上 述症状减轻或消失,因此(+)-生物素在医药、食品添加剂和饲料添加剂等领域得到广泛应 用。
[0005]
(+)-生物素合成路线长、步骤多,现有制备方法均为传统间歇釜式方法,即多步釜式 合成,从起始原料到目标产物的多个合成反应逐步进行,一步反应在间歇式反应釜完成之 后,进行相应的分离、纯化步骤后,再将所获中间体在间歇式反应釜内进行下一步反应, 然后再进行相应的分离、纯化等操作,如此反复进行,直至得到目标产物(+)-生物素。每 个反应包括其相应的分离、纯化等后处理都是一个个步骤独立进行,导致工艺过程损耗大、 收率低、操作繁复、工艺过程效率低、自动化程度低、操作工人数量要求多和工人劳动强 度大等的问题。另外,传统间歇式反应釜本身的分子混合性能及传热传质差等局限,也会 造成间歇釜式合成反应时间长、安全隐患大、物耗高(即单耗高)和能耗高等突出问题。 因此,基于现有制备方法存在的问题,开发一种反应时间短、能耗低、物耗低、工艺过程 效率高以及本质安全的连续化制备方法是本领域技术人员亟需解决的问题。
技术实现要素:
[0006]
本发明的目的在于克服现有技术存在的不足,而提供一种(+)-生物素的全连续流制备 方法,该方法的总制备时间极大缩短,工艺过程的自动化程度和时空效率显著提高,目标 产物的总收率大幅提高,物耗、能耗大幅降低,安全性极大提升,易于工业化应用。
[0007]
本发明提供的(+)-生物素(1)的全连续流制备方法,反应在多级串联的微反应系统 中进行,每级微反应系统包括连通的微反应混合器和微通道反应器,制备的具体步骤为:
[0008]
s1:将环酸酐(2)与联苯类手性丙二醇(3)在有机碱(4)存在下进行不对称开环 反应,制得第一产物二羧酸单酯化合物(5);
[0009]
s2:将s1步骤生成的第一产物二羧酸单酯化合物(5)与硼氢化物(6)进行选择性 还原反应,制得第二产物5-羟甲基-4-羧酸化合物(7);
优选地,所述有机碱(4)为三正丁胺,来源广泛,成本低。
[0025]
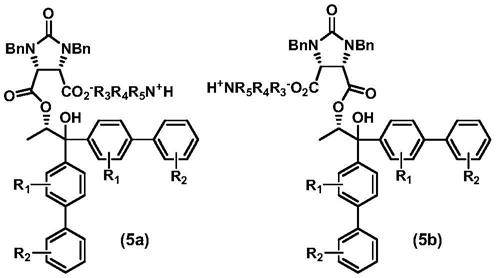
步骤s1中,环酸酐(2)与联苯类手性丙二醇(3)在有机碱(4)存在下进行不对称 开环反应转化为第一产物二羧酸单酯化合物(5),其包括式(5a)和(5b)两个非对映异构 体,它们的结构式如下:
[0026][0027]
作为一种优选的技术方案,步骤s1中,环酸酐(2)与联苯类手性丙二醇(3)在有机 碱(4)存在下进行的不对称开环反应在第一个微通道反应器内进行;环酸酐(2)的溶液 进入第一个微混合器的一个流体入口,联苯类手性丙二醇(3)与有机碱(4)的溶液进入 第一个微混合器的另一个流体入口,第一个微混合器的出口与第一个微通道反应器的一个 流体入口直接连接;环酸酐(2)的溶液与联苯类手性丙二醇(3)及有机碱(4)的溶液 经第一个微混合器混合后紧接着直接进入第一个微通道反应器内进行连续不对称开环反 应。
[0028]
作为一种优选的技术方案,在步骤s1中,控制环酸酐(2)、联苯类手性丙二醇(3) 和有机碱(4)的摩尔比在1:(0.8~3.0):(0.8~3.0)范围内,反应可顺利完成;更优选地, 控制环酸酐(2)、联苯类手性丙二醇(3)与有机碱(4)的摩尔比为1:(0.90~2.5):(0.90~2.5) 是最好的物料比,不但反应可以顺利完成,而且节省物料。
[0029]
作为一种优选的技术方案,用于配制步骤s1中环酸酐(2)的溶液和联苯类手性丙二 醇(3)与有机碱(4)的溶液的溶剂选自苯、甲苯、二甲苯、苯甲醚、氟苯、氯苯、溴苯、 二氯甲烷、三氯甲烷、1,2-二氯乙烷、四氢呋喃、1,4-二氧六环、乙二醇二甲醚、乙醚、 正己烷、环己烷、乙腈、丙酮、n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、二甲基亚砜、 n-甲基吡咯烷酮、环丁砜、1,3-二甲基-2-咪唑啉酮、六甲基磷酰三胺、n-烷基吡啶鎓盐、 1,3-二烷基咪唑鎓盐中的一种,或其中多种的混合溶剂;更优选地,步骤s1中各反应物 溶液配置都采用四氢呋喃或甲苯可以获得更佳的反应效果。
[0030]
作为一种优选的技术方案,在步骤s1中,第一个微混合器为静态混合器、t型微混合 器、y型微混合器、同轴流动微混合器(coaxial flow micromixer)和流动聚焦微混合器 (flow-focusing micromixer)等中的任何一种。
[0031]
作为一种优选的技术方案,在步骤s1中,控制所述第一个微混合器内的温度在-20~80℃ 范围内。
[0032]
作为一种优选的技术方案,在步骤s1中,控制所述第一个微通道反应器内的温度在
ꢀ‑
20~80℃范围内,反应可顺利完成;更优选地,将第一个微通道反应器内的温度控制在
ꢀ‑
15~60℃范围内,反应效果好,能耗更低。
[0033]
作为一种优选的技术方案,在步骤s1中,所述的第一个微通道反应器是管式微通道 反应器或板式微通道反应器;所述管式微通道反应器的内径为100微米~10毫米;更优
选 地,所述管式微通道反应器的内径为120微米~5.35毫米;所述板式微通道反应器包括从 上至下依次设置的第一换热层、反应层和第二换热层;所述反应层设有反应流体通道;所 述反应流体通道的水力直径为100微米~10毫米;更优选地,所述反应流体通道的水力直 径为120微米~5.35毫米。
[0034]
作为一种优选的技术方案,在步骤s1中,所述的第一个微通道反应器是振荡式微通道 反应器;更优选地,所述振荡式微通道反应器是科弗洛连续流反应器(coflore agitated cell reactor,英国am technology公司)。
[0035]
作为一种优选的技术方案,在步骤s1中,控制进入第一个微混合器的两股反应液的总 流量,使得从第一个微混合器流出的混合反应物料在第一个微通道反应器内的停留时间在 1~40分钟范围内,反应可顺利完成。
[0036]
步骤s2中,所述硼氢化物(6)选自硼氢化锂、硼氢化钠、硼氢化钾、硼氢化钙中的 任一种;优选地,所述硼氢化物(6)最好为硼氢化锂,其化学性质稳定,反应效果更佳, 且价廉易得。
[0037]
步骤s2中,将s1步骤生成的第一产物二羧酸单酯化合物(5)与硼氢化物(6)进行 选择性还原反应转化为第二产物5-羟甲基-4-羧酸化合物(7)。
[0038]
作为一种优选的技术方案,步骤s2中的第一产物二羧酸单酯化合物(5)与硼氢化物 (6)进行的选择性还原反应在第二个微通道反应器内进行;第一个微通道反应器的出口 与第二个微混合器的一个流体入口连接,从第一个微通道反应器流出的反应液直接进入第 二个微混合器的这个流体入口,硼氢化物(6)溶液进入第二个微混合器的另一个流体入 口,第二个微混合器的出口与第二个微通道反应器的一个流体入口直接连接;从第一个微 通道反应器流出的反应液与硼氢化物(6)溶液经第二个微混合器混合后紧接着直接进入 第二个微通道反应器内进行连续选择性还原反应。
[0039]
作为一种优选的技术方案,步骤s2中,用于配制硼氢化物(6)溶液的溶剂选自苯、 甲苯、二甲苯、苯甲醚、氟苯、氯苯、溴苯、二氯甲烷、三氯甲烷、1,2-二氯乙烷、四 氢呋喃、1,4-二氧六环、乙二醇二甲醚、乙醚、正己烷、环己烷、乙腈、丙酮、n,n-二 甲基甲酰胺、n,n-二甲基乙酰胺、二甲基亚砜、n-甲基吡咯烷酮、环丁砜、1,3-二甲基
ꢀ‑
2-咪唑啉酮、六甲基磷酰三胺、n-烷基吡啶鎓盐、1,3-二烷基咪唑鎓盐中的一种,或其 中多种的混合溶剂;更优选地,所述有机溶剂为四氢呋喃或甲苯更好,来源广泛,成本更 低。
[0040]
作为一种优选的技术方案,步骤s2中,所述第二个微混合器为静态混合器、t型微混 合器、y型微混合器、同轴流动微混合器(coaxial flow micromixer)和流动聚焦微混合 器(flow-focusing micromixer)等中的任何一种。
[0041]
作为一种优选的技术方案,步骤s2中,控制硼氢化物(6)与第一产物二羧酸单酯化 合物(5)的摩尔比在(1~8):1范围内,反应可顺利完成;更优选地,控制硼氢化物(6) 与s1步骤生成的第一产物二羧酸单酯化合物(5)的摩尔比为(1.02~5.0):1更好,不但 反应可顺利完成,而且节省物料。
[0042]
作为一种优选的技术方案,步骤s2中,控制第二个微混合器内的温度在0~100℃范 围内。
[0043]
作为一种优选的技术方案,步骤s2中,控制第二个微通道反应器内的温度在0~100℃ 范围内,反应可顺利完成;更优选地,将第二个微通道反应器内的温度控制在0~80
℃ 范围内,反应效果更好,能耗更低。
[0044]
作为一种优选的技术方案,步骤s2中,所述第二个微通道反应器是管式微通道反应 器或板式微通道反应器;所述管式微通道反应器的内径为100微米~10毫米;更优选地, 所述管式微通道反应器的内径为120微米~5.35毫米;所述板式微通道反应器包括从上至 下依次设置的第一换热层、反应层和第二换热层;所述反应层设有反应流体通道;所述反 应流体通道的水力直径为100微米~10毫米;更优选地,所述反应流体通道的水力直径为 120微米~5.35毫米。
[0045]
作为一种优选的技术方案,步骤s2中,第二个微通道反应器的出口与第二a微混合 器的一个流体入口连接,第二a微混合器的另一个流体入口通入水,流出第二个微通道反 应器的反应混合液与水在第二a微混合器内混合,之后进入第一个连续液液萃取器,用乙 酸乙酯或甲苯萃取后,有机相从第一个连续液液萃取器的有机相出口流出,水相从第一个 连续液液萃取器的水相出口流出,收集有机相可以回收所述联苯类手性丙二醇(3)重复 利用。
[0046]
步骤s3中,步骤s2生成的第二产物5-羟甲基-4-羧酸化合物(7)与无机矿酸(8) 溶液进行环合反应转化为第三产物(3as,6ar)内酯(9),所述第三产物(3as,6ar) 内酯(9)的结构式为:
[0047][0048]
作为一种优选的技术方案,步骤s2中的第二产物5-羟甲基-4-羧酸化合物(7)与无机 矿酸(8)进行连续环合反应在第三个微通道反应器内进行;第一个连续液液萃取器的水 相出口与第三个微混合器的一个流体入口连接,从第一个连续液液萃取器的水相出口流出 的反应液直接进入第三个微混合器的这个流体入口,无机矿酸(8)溶液进入第三个微混 合器的另一个流体入口,第三个微混合器的出口与第三个微通道反应器的一个流体入口直 接连接;从第二个微通道反应器流出的反应液与无机矿酸(8)溶液经第三个微混合器混 合后紧接着直接进入第三个微通道反应器内进行连续环合反应。
[0049]
步骤s3中,所述无机矿酸(8)选自盐酸、硫酸、硝酸和磷酸等中的任一种;优选地, 所述无机矿酸(8)为盐酸,反应效果更佳。
[0050]
步骤s3中,所述无机矿酸(8)溶液为无机矿酸(8)溶于水配制成的溶液。
[0051]
作为一种优选的技术方案,步骤s3中,所述的第三个微混合器为静态混合器、t型微 混合器、y型微混合器、同轴流动微混合器(coaxial flow micromixer)和流动聚焦微混 合器(flow-focusing micromixer)等中的任何一种。
[0052]
作为一种优选的技术方案,步骤s3中,控制无机矿酸(8)与第二产物5-羟甲基-4
‑ꢀ
羧酸化合物(7)的摩尔比在(1~50):1范围内,反应可顺利完成;更优选地,所述无机 矿酸(8)与步骤s1中的环酸酐(2)的摩尔比为(1~30):1更好,不但反应可顺利完成, 而且节省物料。
[0053]
作为一种优选的技术方案,步骤s3中,控制第三个微混合器内的温度在-10~120
℃ 范围内。
[0054]
作为一种优选的技术方案,步骤s3中,控制第三个微通道反应器内的温度在10~150℃ 范围内,反应可顺利完成;更优选地,将所述第三个微通道反应器内的温度控制在20~120℃ 范围内,反应效果更好,能耗更低。
[0055]
作为一种优选的技术方案,步骤s3中,所述的第三个微通道反应器是管式微通道反 应器或板式微通道反应器;所述管式微通道反应器的内径为100微米~10毫米;更优选地, 所述管式微通道反应器的内径为120微米~5.35毫米;所述板式微通道反应器包括从上至 下依次设置的第一换热层、反应层和第二换热层;所述反应层设有反应流体通道;所述反 应流体通道的水力直径为100微米~10毫米;更优选地,所述反应流体通道的水力直径为 120微米~5.35毫米。
[0056]
作为一种优选的技术方案,步骤s3中,第三个微通道反应器的出口与第二个连续液 液萃取器的一个流体入口连接,流出第三个微通道反应器的反应混合液紧接着进入第二个 连续液液萃取器,用乙酸乙酯或甲苯萃取后,有机相从第二个连续液液萃取器的有机相出 口流出,水相从第二个连续液液萃取器的水相出口流出。第二个连续液液萃取器的有机相 出口与第一个连续提浓器的入口连接,从第二个连续液液萃取器的有机相出口流出的反应 液进入第一个连续提浓器进行连续提浓。
[0057]
步骤s4中,所述硫代试剂(10)的结构式为:
[0058][0059]
式(10)中,r6为c1~c6烷基、c3~c6环烷基,x为氧或硫,q为钾或钠。
[0060]
步骤s4中,(3as,6ar)内酯(9)与硫代试剂(10)进行硫代反应转化为第四产物 (3as,6ar)硫内酯(11),所述第四产物(3as,6ar)硫内酯(11)的结构式为:
[0061][0062]
作为一种优选的技术方案,步骤s4中的(3as,6ar)内酯(9)与硫代试剂(10)的 硫代反应在第四个微通道反应器内进行;第一个连续提浓器的出口与第四个微混合器的一 个流体入口连接,从第一个连续提浓器的出口流出的反应液直接进入第四个微混合器的这 个流体入口,硫代试剂(10)的溶液进入第四个微混合器的另一个流体入口,第四个微混 合器的出口与第四个微通道反应器的一个流体入口直接连接,第四个微通道反应器的出口 与第一个背压阀的入口连接;从第一个连续提浓器的出口流出的反应液和硫代试剂(10) 的溶液经第四个微混合器混合后紧接着直接进入第四个微通道反应器内进行连续硫代反 应;流出第四个微通道反应器的反应液接着进入第一个背压阀。
[0063]
作为一种优选的技术方案,步骤s4中,用于配制硫代试剂(10)的溶液的溶剂为n, n-二甲基甲酰胺、n,n-二甲基乙酰胺、n,n-甲基吡咯烷酮、环丁砜、二氯亚溶剂等中的 任何
接进入第五个微通道反应器的这个流体入口,含钯催化剂的锌试剂(12)溶液进入第五个 微混合器的另一个流体入口,第五个微通道反应器的出口与第二个背压阀的入口连接;从 第一个背压阀流出的反应液与含钯催化剂的锌试剂溶液经第五个微混合器混合后形成的 反应混合液紧接着直接进入第五个微通道反应器内进行连续fukuyama偶联反应;流出第五 个微通道反应器的反应混合液紧接着直接进入第二个背压阀。
[0077]
作为一种优选的技术方案,步骤s5中,所述钯催化剂为醋酸钯、二(三苯基膦)二 氯化钯、钯/碳、纳米钯和氢氧化钯/碳等中的任一种。
[0078]
作为一种优选的技术方案,步骤s5中,用于配制含钯催化剂的锌试剂溶液的溶剂为n, n-二甲基甲酰胺、n,n-二甲基乙酰胺、n,n-甲基吡咯烷酮、环丁砜、二氯亚溶剂等中的 任何一种;更优选地,所述有机溶剂为n,n-二甲基甲酰胺或n,n-二甲基乙酰胺。
[0079]
作为一种优选的技术方案,步骤s5中,控制(3as,6ar)硫内酯(11)与所述钯催 化剂的摩尔比为1:(0.001~0.500)。
[0080]
作为一种优选的技术方案,步骤s5中,第五个微混合器为静态混合器、t型微混合器、 y型微混合器、同轴流动微混合器(coaxial flow micromixer)和流动聚焦微混合器 (flow-focusing micromixer)等中的任一种。
[0081]
作为一种优选的技术方案,步骤s5中,控制(3as,6ar)硫内酯(11)与锌试剂(12) 的摩尔比在1:(1.0~8.0)范围内。
[0082]
作为一种优选的技术方案,步骤s5中,第五个微混合器内的温度控制在5~160℃范 围内;更优选地,所述第五个微混合器内的温度控制在30~155℃范围内。
[0083]
作为一种优选的技术方案,步骤s5中,第五个微通道反应器内的温度控制在5~160℃ 范围内;更优选地,所述第五个微通道反应器内的温度控制在30~155℃范围内。
[0084]
作为一种优选的技术方案,步骤s5中,所述的第五个微通道反应器是管式微通道反 应器或板式微通道反应器;所述管式微通道反应器的内径为100微米~10毫米;更优选地, 所述管式微通道反应器的内径为120微米~5.35毫米;所述板式微通道反应器包括从上至 下依次设置的第一换热层、反应层和第二换热层;所述反应层设有反应流体通道;所述反 应流体通道的水力直径为100微米~10毫米;更优选地,所述反应流体通道的水力直径为 120微米~5.35毫米。
[0085]
作为一种优选的技术方案,步骤s5中,控制混合反应物料在第五个微通道反应器内 的停留时间为0.5~30分钟;更优选地,控制混合反应物料在第五个微通道反应器内的停 留时间为1~27分钟。
[0086]
作为一种优选的技术方案,步骤s6中,第五产物羟基戊酸酯化合物(13)在无机矿 酸(14)溶液作用下进行消除反应转化为第六产物烯基戊酸酯化合物(15),所述第六产 物烯基戊酸酯化合物(15)的结构式为:
[0087][0088]
步骤s6中,所述无机矿酸(14)为硫酸、盐酸、硼酸、磷酸、碳酸和硝酸等中的任 一
种。
[0089]
作为一种优选的技术方案,步骤s6中,第五产物羟基戊酸酯化合物(13)在无机矿酸 (14)溶液作用下的消除反应在第六个微通道反应器内进行;第二个背压阀的出口与第六 个微混合器的一个流体入口连接,从第二个背压阀的出口流出的反应液直接进入第六个微 混合器的这个流体入口,无机矿酸(14)溶液进入第六个微混合器的另一个流体入口;第 六个微混合器的出口与第六个微通道反应器的一个流体入口直接连接,从第六个微混合器 流出的反应混合液直接进入第六个微通道反应器的这个流体入口;从第二个背压阀的出口 流出的反应混合液与无机矿酸(14)溶液经第六个微混合器混合后紧接着直接进入第六个 微通道反应器内进行连续消除反应。
[0090]
作为一种优选的技术方案,步骤s6中,第六个微通道反应器的出口与第一个连续液液 分离器的一个流体入口连接,流出第六个微通道反应器的反应混合液紧接着直接进入第一 个连续液液分离器,水相从第一个连续液液分离器的水相出口流出,有机相从第一个连续 液液分离器的有机相出口流出。
[0091]
作为一种优选的技术方案,步骤s6中,所述无机矿酸(14)溶液为无机矿酸(14) 溶于水制成的溶液。
[0092]
作为一种优选的技术方案,步骤s6中,所述的第七个微混合器为静态混合器、t型微 混合器、y型微混合器、同轴流动微混合器(coaxial flow micromixer)和流动聚焦微混 合器(flow-focusing micromixer)等中的任一种。
[0093]
作为一种优选的技术方案,步骤s6中,所述的第六个微混合器内的温度控制在5~100℃ 范围内;更优选地,第六个微混合器的温度控制在10~90℃范围内。
[0094]
作为一种优选的技术方案,步骤s6中,控制第五产物羟基戊酸酯化合物(13)与无 机矿酸(14)的摩尔比控制在1:(1~50)范围内。
[0095]
作为一种优选的技术方案,步骤s6中,所述的第六个微通道反应器是管式微通道反 应器或板式微通道反应器;所述管式微通道反应器的内径为100微米~10毫米;更优选地, 所述管式微通道反应器的内径为120微米~5.35毫米;所述板式微通道反应器包括从上至 下依次设置的第一换热层、反应层和第二换热层;所述反应层设有反应流体通道;所述反 应流体通道的水力直径为100微米~10毫米;更优选地,所述反应流体通道的水力直径为 120微米~5.35毫米。
[0096]
作为一种优选的技术方案,步骤s6中,控制混合反应物料在第六个微通道反应器内 的停留时间为0.5~30分钟;更优选地,控制混合反应物料在第六个微通道反应器内的停 留时间为1~27分钟。
[0097]
作为一种优选的技术方案,步骤s6中,所述的第六个微通道反应器内的温度控制在 5~100℃范围内;更优选地,第六个微通道反应器内的温度控制在10~90℃范围内。
[0098]
步骤s7中,第六产物烯基戊酸酯化合物(15)在钯/碳催化剂作用下进行加氢还原转 化为第七产物戊酸酯(16),所述第七产物戊酸酯(16)的结构式为:
[0099]
[0100]
作为一种优选的技术方案,步骤s7中,第六产物烯基戊酸酯化合物(15)在钯/碳催 化剂作用下的加氢还原反应在第七个微通道反应器内进行;第一个连续液液分离器的有机 相出口与第七个微混合器的一个流体入口连接,从第一个连续液液分离器的有机相出口流 出的反应液直接进入第七个微混合器的这个流体入口,氢气进入第七个微混合器的另一个 流体入口;第七个微混合器的出口与第七个微通道反应器的一个流体入口直接连接,从第 七个微混合器流出的反应混合液直接进入第七个微通道反应器;从第一个连续液液分离器 的有机相出口流出的反应液与氢气经第七个微混合器混合后紧接着直接进入第七个微通 道反应器内进行连续加氢还原反应;第七个微通道反应器的出口与一个缓冲罐连接,缓冲 罐底部装有带阀门的液体出口,顶部有与氮气管路连接的第一接口,与第七个微通道反应 器的出口连接的第二接口,以及一个与背压阀连接的第三接口;所述第一接口接入氮气, 用于给所述缓冲罐提供压力,接入氮气的压力可调范围为0.1~2.0mpa,所述第三接口与 第三个背压阀连接;所述第三个背压阀的背压范围为0.1~1.5mpa;接入氮气的压力值 要比第三个背压阀设置的背压值大0.2~1.0mpa;所述第二接口与第七个微通道反应器的 出口连接。
[0101]
作为一种优选的技术方案,步骤s7中,所述钯/碳催化剂为负载量为0.5~30%的钯碳 (pd/c)催化剂或负载量为0.5~30%的氢氧化钯碳(pd(oh)2/c)催化剂;更优选地,所述 钯/碳催化剂为负载量为0.5~30%的钯碳(pd/c)催化剂或负载量为0.5~30%的氢氧化钯碳 (pd(oh)2/c)催化剂与惰性固体介质颗粒物(如石英砂、硅藻土、玻璃珠、硅胶等)拌匀 混合后形成的混合物。
[0102]
作为一种优选的技术方案,步骤s7中,控制第六产物烯基戊酸酯化合物(15)与氢 气的摩尔比在1:(0.8~50.0)范围内。
[0103]
作为一种优选的技术方案,步骤s7中,所述第七个微混合器内的温度控制在10~120℃。
[0104]
作为一种优选的技术方案,步骤s7中,所述第七个微通道反应器内的温度控制在 15~120℃范围内;
[0105]
作为一种优选的技术方案,步骤s7中,控制混合反应物料在第七个微通道反应器内 的停留时间在0.1~30分钟范围内。
[0106]
作为一种优选的技术方案,步骤s7中,所述第七个微混合器为静态混合器、t型微混 合器、y型微混合器、同轴流动微混合器(coaxial flow micromixer)或流动聚焦微混合 器(flow-focusing micromixer)中的任一种。
[0107]
作为一种优选的技术方案,步骤s7中,所述第七个微通道反应器是填充有钯/碳催化 剂的微型固定床反应器;所述微型固定床反应器的内径为1毫米~100毫米,更优选为1.5 毫米~70毫米。
[0108]
步骤s8中,第七产物戊酸酯(16)在无机碱(17)溶液作用下进行水解反应转化为 第八产物戊酸盐(18),所述第八产物戊酸盐(18)的结构式为:
[0109][0110]
其中z为钠或钾离子。
[0111]
作为一种优选的技术方案,步骤s8中,第七产物戊酸酯(16)在无机碱(17)溶液作 用下进行的水解反应在第八个微通道反应器内进行;所述缓冲罐内的反应液输送到第八个 微混合器的一个流体入口,无机碱(17)溶液进入第八个微混合器的另一个流体入口;第 八个微混合器的出口与第八个微通道反应器的一个流体入口直接连接,从第八个微混合器 流出的反应混合液直接进入第八个微通道反应器内进行连续水解反应。
[0112]
作为一种优选的技术方案,步骤s8中,所述无机碱(17)为氢氧化钠、氢氧化钾、 氢氧化锂、碳酸钠、碳酸钾和碳酸锂等中的任一种;所述无机碱(17)溶液为无机碱(17) 溶于水制成的溶液。
[0113]
作为一种优选的技术方案,步骤s8中,第八个微通道反应器的出口与第二个连续液液 分离器的一个流体入口连接,流出第八个微通道反应器的反应混合液紧接着直接进入第二 个连续液液分离器,水相从第二个连续液液分离器的一个水相出口流出,有机相从第二个 连续液液分离器的有机相出口流出。
[0114]
作为一种优选的技术方案,步骤s8中,所述第八个微混合器为静态混合器、t型微混 合器、y型微混合器、同轴流动微混合器(coaxial flow micromixer)或流动聚焦微混合 器(flow-focusing micromixer)中的任一种。
[0115]
作为一种优选的技术方案,步骤s8中,控制戊酸酯(16)与无机碱(17)的摩尔比 为1:(0.75~30)。
[0116]
作为一种优选的技术方案,步骤s8中,所述第八个微混合器内的温度控制在2~120℃ 范围内。
[0117]
作为一种优选的技术方案,步骤s8中,所述第八个微通道反应器内的温度分别控制 在15~150℃范围内。
[0118]
作为一种优选的技术方案,步骤s8中,所述第八个微通道反应器是管式微通道反应 器,或板式微通道反应器;所述管式微通道反应器的内径为100微米~50毫米,更优选为 120微米~30毫米;所述板式微通道反应器的反应流体通道的水力直径为100微米~50毫米, 更优选为120微米~30毫米。
[0119]
作为一种优选的技术方案,步骤s8中,控制混合反应物料在第八微通道反应器内的 停留时间控制在0.1~30分钟范围内。
[0120]
步骤s9中,第八产物戊酸盐(18)在无机矿酸(19)溶液作用下进行脱苄反应转化 为目标产物(+)-生物素(1)。
[0121]
作为一种优选的技术方案,步骤s8中,第八产物戊酸盐(18)在无机矿酸(19)溶液 作用下的脱苄反应在第九个微通道反应器内进行;从第二个连续液液分离器的水相出口流 出的反应液进入第九个微混合器的一个流体入口,无机矿酸(19)溶液进入第九个微混合 器的另一个流体入口;第九个微混合器的出口与第九个微通道反应器的一个流体入口连
接, 从第九个微混合器流出的反应混合液直接进入第九个微通道反应器内进行连续脱苄反应。
[0122]
作为一种优选的技术方案,步骤s9中,所述无机矿酸(19)为溴化氢或氯化氢;所 述无机矿酸(19)溶液为无机矿酸(19)溶于水制成的溶液。
[0123]
作为一种优选的技术方案,步骤s9中,控制戊酸盐(18)与无机矿酸(19)的摩尔 比为1:(0.75~30)。
[0124]
作为一种优选的技术方案,步骤s9中,所述第九个微混合器内的温度控制在-10~25℃。
[0125]
作为一种优选的技术方案,步骤s9中,所述第九个微通道反应器内的温度控制在 25~200℃;
[0126]
作为一种优选的技术方案,步骤s9中,控制混合反应物料在第九个微通道反应器内 的停留时间控制在0.1~30分钟。
[0127]
作为一种优选的技术方案,步骤s9中,所述第九个微混合器为静态混合器、t型微混 合器、y型微混合器、同轴流动微混合器(coaxial flow micromixer)或流动聚焦微混合 器(flow-focusing micromixer)中的任一种。
[0128]
作为一种优选的技术方案,步骤s9中,所述第九个微通道反应器是管式微通道反应 器,或板式微通道反应器;所述管式微通道反应器的内径为100微米~50毫米,更优选为 120微米~30毫米;所述板式微通道反应器的反应流体通道的水力直径为100微米~50毫米, 更优选为120微米~30毫米。
[0129]
作为一种优选的技术方案,步骤s9中,所述微通道反应器是微波流动化学反应器, 其反应流体通道的水力直径为100微米~50毫米,更优选为120微米~30毫米。
[0130]
作为一种优选的技术方案,步骤s9中,所述第九个微通道反应器的出口与第十个微 通道反应器的一个流体入口连接,流出第九个微通道反应器出口的反应液直接进入第十个 微通道反应器。
[0131]
作为一种优选的技术方案,步骤s9中,所述第十个微通道反应器是管式微通道反应 器,或板式微通道反应器;所述管式微通道反应器的内径为1毫米~10毫米;所述板式微 通道反应器的反应流体通道的水力直径为1毫米~10毫米。
[0132]
作为一种优选的技术方案,控制第十个微通道反应器内的温度在-35~10℃范围内, 目标产物可以结晶析出,收集流出第十个微通道反应器的反应混合液过滤即得目标产物(+)
ꢀ‑
生物素。
[0133]
本发明有益效果:本发明的连续制备(+)-生物素的方法,相比现有的多步间歇釜式 合成方法具有以下优势:
[0134]
1.实现从原料到目标产物(+)-生物素的连续九步合成,工艺过程连续不间断进行, 自动化程度高,中间无需外部干预,时空效率高,大幅减少操作工人数量和劳动强度,显 著降低生产成本。
[0135]
2.高温高压和加氢反应等危险反应过程都在微通道反应器的反应流体通道内完成, 反应流体通道总容积小,使得在线持液量小,整个工艺过程本质安全。
[0136]
3.九步连续反应,总制备时间大大缩短,从九步间歇釜式合成需要两周左右的时间 (包括反应以及每步相应的后处理时间)缩短到1小时以内。
[0137]
4.反应过程的物料混合、传质与反应过程在微混合器和微通道反应器的反应流体通 道内完成,无需搅拌装置,大幅减小工艺过程总能耗。
[0138]
5.通过微通道反应器强化反应过程,提高每步反应的收率,以及通过将多步反应串 联,减少中间的后处理过程,减少物料损失,使得九步反应的总收率从间歇釜式方法的 30~35%左右提高至48.7%以上。
具体实施方式
[0139]
为详细说明技术方案的技术内容、构造特征、所实现目的及效果,以下结合具体实施 例作进一步说明。本实施例在以本发明技术方案为前提下进行实施,给出了详细的实施方 式和具体的操作过程,但本发明的保护范围不限于下述的实施例。
[0140]
实施例1
[0141]
用进料泵将环酸酐(2)的四氢呋喃溶液(0.1m)输送到第一个微混合器(内径0.8 毫米的t型微混合器)的一个流体入口,联苯类手性丙二醇(3,其中r1为氢,r2为氢) 及三正丁胺的四氢呋喃溶液输送到第一个微混合器的另一个流体入口,精确控制两股反应 液的流量比,使得环酸酐(2)、联苯类手性丙二醇(3,其中r1为氢,r2为氢)与三正丁 胺的摩尔比为1:1.05:1,第一个微混合器内的温度控制为0℃。从第一个微混合器出 口流出的反应混合液紧接着直接进入第一个微通道反应器(聚四氟乙烯管,内径为0.8mm, 体积1.5ml),第一个微通道反应器内的温度控制为25℃,反应7.5分钟后(即混合反 应物料在第一个微通道反应器内的停留时间为7.5分钟),混合反应物料从第一个微通道 反应器内的出口流出。
[0142]
从第一个微通道反应器出口流出的混合反应物料进入第二个微混合器(内径0.8毫米 的t型微混合器)的一个流体入口,用进料泵将硼氢化锂的四氢呋喃溶液(0.8m)输送 至第二个微混合器的另一个流体入口,调节硼氢化锂的四氢呋喃溶液的流量,使得硼氢化 锂与环酸酐(2)的摩尔比为1.5:1,第二个微混合器内的温度控制为25℃;从第二个 微混合器的出口流出的混合反应物料紧接着直接进入第二个微通道反应器(聚四氟乙烯管, 内径为0.8mm,体积3.0ml),第二个微通道反应器内的温度控制为45℃,反应2.5分 钟后(即混合反应物料在第二个微通道反应器的停留时间为2.5分钟),混合反应物料从 第二个微通道反应器的出口流出。从第二个微通道反应器出口流出的反应混合液与水在第 二a微混合器内混合后,进入第一个连续液液萃取器,用乙酸乙酯连续萃取,有机相从第 一个连续液液萃取器的有机相出口流出,水相从第一个连续液液萃取器的水相出口流出。
[0143]
从第一个连续液液萃取器水相出口流出的反应液直接进入第三个微混合器(内径0.8 毫米的t型微混合器)的一个流体入口,用进料泵将盐酸水溶液(1.0m)输送至第三个 微混合器的另一个流体入口,调节盐酸水溶液的流量,使得氯化氢与环酸酐(2)的摩尔 比为5:1,第三个微混合器内的温度控制为60℃,从第一个连续液液萃取器分出的水相 与盐酸水溶液在第三个微混合器内混合;从第三个微混合器的出口流出的混合反应物料紧 接着直接进入第三个微通道反应器(聚四氟乙烯管,内径为0.6mm,体积3.0ml),第三 个微通道反应器内的温度控制为60℃,反应1分钟后(即混合反应物料在微通道反应器 内的停留时间为1分钟),混合反应物料从第三个微通道反应器的出口流出。从第三个微 通道反应器的出口流出的反应混合液紧接着进入第二个连续液液萃取器,用甲苯萃取,有 机相从第二个连续液液萃取器的有机相出口流出,水相从第二个连续液液萃取器的水相出 口流出。从
第二个连续液液萃取器的有机相出口流出的反应液接着进入第一个连续提浓器 进行提浓。
[0144]
从第一个连续提浓器出口流出的反应液进入第四个微混合器(内径0.8毫米的t型微混 合器)的一个流体入口,用进料泵将硫代乙酸钾溶液(硫代试剂溶液,0.3m)输送至第 四个微混合器的另一个流体入口,调节硫代乙酸钾溶液的流量,使得硫代乙酸甲与环酸酐 (2)的摩尔比为1.2:1,第四个微混合器内的温度控制为145℃,从第三个微通道反应 器出口流出的混合反应物料经连续提浓后与硫代乙酸钾的二甲基甲酰胺溶液在第四个微 混合器内混合;从第四个微混合器的出口流出的混合反应物料紧接着直接进入第四个微通 道反应器(聚四氟乙烯管,内径为0.8mm,体积4.0ml),第四个微通道反应器内的温度 控制为145℃,反应10分钟后(即混合反应物料在微通道反应器内的停留时间为10分钟), 混合反应物料从第四个微通道反应器的出口流出后进入第一个背压阀的入口,然后丛第一 个背压阀的出口流出。
[0145]
从第一个背压阀的出口流出的混合反应物料直接进入第五个微混合器(内径0.8毫米 的t型微混合器)的一个流体入口,用进料泵将含5mol%醋酸钯的(5-乙氧基-5-氧代戊 基)溴化锌四氢呋喃溶液(锌试剂溶液,0.25m)输送至第五个微混合器的另一个流体入 口,控制环酸酐(2)与(5-乙氧基-5-氧代戊基)溴化锌的摩尔比为1:1.05,第五个微 混合器内的温度控制为105℃,从第五个微混合器的出口流出的混合反应物料紧接着直接 进入第五个微通道反应器(聚四氟乙烯管,内径为0.6mm,体积4.0ml),反应物料在其 内进行fukuyama偶联反应,第五个微通道反应器内的温度控制为105℃,反应10分钟后 (即混合反应物料在微通道反应器内的停留时间为10分钟),混合反应物料从第五个微通 道反应器的出口流出后进入第二个背压阀的入口,然后丛第二个背压阀的出口流出。
[0146]
从第二个背压阀的出口流出的混合反应物料直接进入第六个微混合器(内径0.8毫米 的t型微混合器)的一个流体入口,用进料泵将盐酸水溶液(1.0m)输送至第六个微混 合器的另一个流体入口,调节盐酸水溶液的流量,使得环酸酐(2)与氯化氢的摩尔比为1: 8,第六个微混合器内的温度控制为45℃,从第二个背压阀的出口流出的混合反应物料与 盐酸水溶液在第六个微混合器内混合;从第六个微混合器的出口流出的混合反应物料紧接 着直接进入第六个微通道反应器(聚四氟乙烯管,内径为0.8mm,体积3.0ml),反应物 料在其内进行消除反应,第六个微通道反应器内的温度控制为45℃,反应10分钟后(即 混合反应物料在微通道反应器内的停留时间为10分钟),混合反应物料从第六个微通道反 应器的出口流出。从第六个微通道反应器出口流出的反应混合液进入第一个连续液液分离 器,有机相从第一个连续液液分离器的有机相出口流出,水相从第一个连续液液分离器的 水相出口流出。
[0147]
从第一个连续液液分离器的有机相出口流出的反应液直接进入第七个微混合器(内径 0.8毫米的t型微混合器)的一个流体入口,将氢气输送到第七个微混合器的另一个流体 入口,调节氢气的流量,使得环酸酐(2)与氢气的摩尔比为1:3,第七个微混合器内的 温度控制为25℃,从第七个微混合器的出口流出的混合反应物料紧接着直接进入第七个 微通道反应器(长度为20cm,内径为1cm的微型固定床反应器,填充有5.8克负载量 为10%的钯/碳催化剂与14.2克石英砂的均匀混合物,填充钯/碳催化剂后的微型固定床反 应器内的反应体积约4.0ml),控制的微型固定床反应器内的温度控制为25℃,反应2 分钟后(即
混合反应物料在微通道反应器内的停留时间为2分钟),混合反应物料从第七 个微通道反应器的出口流出;从第七个微通道反应器的出口流出的反应混合液进入缓冲罐, 与缓冲罐顶部连接的第三个背压阀的背压值设定为1.0mpa,缓冲罐顶部接入的氮气的压 力调节为1.5mpa。
[0148]
将缓冲罐内的反应液用进料泵输送到第八个微混合器(内径0.8毫米的t型微混合器) 的一个流体入口,用进料泵将氢氧化钠水溶液(1.0m)输送到第八个微混合器的另一个 流体入口,调节氢氧化钠水溶液的流量,使得环酸酐(2)与氢氧化钠水溶液的摩尔比为1: 1.5,第八个微混合器内的温度控制为45℃,从第八个微混合器的出口流出的混合反应物 料紧接着直接进入第八个微通道反应器,控制第八个微通道反应器内的温度控制为25℃, 反应5.0分钟后(即混合反应物料在微通道反应器内的停留时间为5.0分钟),混合反应 物料从第八个微通道反应器的出口流出。从第八个微通道反应器的出口流出的反应混合液 进入第二个连续液液分离器,有机相从第二个连续液液分离器的有机相出口流出,水相从 第二个连续液液分离器的水相出口流出。
[0149]
从第二个连续液液分离器的水相出口流出的反应物料直接进入第九个微混合器(内径 0.8毫米的t型微混合器)的一个流体入口,用进料泵将47%hbr水溶液输送到第九个微混 合器的另一个流体入口,控制环酸酐(2)与hbr的摩尔比为1:5,控制第九个微混合器 内的温度为5℃,从第九个微混合器的出口流出的混合反应物料紧接着直接进入第九个微 通道反应器(第九个微通道反应器为微波连续流反应器,反应流体通道的水力直径为0.8 毫米,反应体积为8.6毫升),第九个微通道反应器内的温度控制为145℃,反应10分钟 后(即混合反应物料在微通道反应器内的停留时间为10分钟),混合反应物料从第九个微 通道反应器的出口流出。
[0150]
从第九个微通道反应器的出口流出的混合反应物料直接进入第十个微通道反应器(内 径为3.2毫米的聚四氟管,反应体积为20毫升),控制微通道反应器内的温度控制为-10℃, 收集从第十个微通道反应器内流出的反应混合物料,过滤即得目标产物(+)-生物素。经 分析,目标产物(+)-生物素总收率为48%,对映选择性100%。
[0151]
实施例2
[0152]
本实施例与实施例1相同,唯一不同之处是本实施例中控制第四个微通道反应器内的 温度为160℃。本实施例目标产物(+)-生物素总收率为49.6%,对映选择性100%。
[0153]
实施例3
[0154]
本实施例与实施例1相同,唯一不同之处是本实施例中控制第五个微通道反应器内的 温度为120℃。本实施例目标产物(+)-生物素总收率为50.2%,对映选择性100%。
[0155]
实施例4
[0156]
本实施例与实施例1相同,唯一不同之处是本实施例中控制第九个微通道反应器内的 温度为155℃。本实施例目标产物(+)-生物素总收率为49.8%,对映选择性100%。
[0157]
实施例5
[0158]
本实施例与实施例1相同,唯一不同之处是本实施例中第九个微通道反应器是普通聚 四氟管(内径为0.8毫米,反应体积为8.6毫升),采用油浴加热而不是微波连续流反应 器。本实施例目标产物(+)-生物素总收率为0%。
[0159]
最后所应当说明的是,以上实施例仅用以说明本发明的技术方案而非对本发明保
护范 围的限制,尽管参照较佳实施例对本发明作了详细说明,本领域的普通技术人员应当理解, 可以对本发明的技术方案进行修改或者等同替换,而不脱离本发明技术方案的实质和范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1