一种噻唑并吡啶环联三氮唑类化合物及其制备方法和应用
1.本发明属于抑制剂制备技术领域,涉及一种噻唑并吡啶环联三氮唑类化合物及其制备方法和应用。
背景技术:
2.慢性粒细胞白血病(cml)是一种发生于骨髓造血干细胞的恶性克隆增生性疾病,在成年白血病患者中所占比例高达15%~20%,其特征是cml患者体内能检测出ph染色体。ph染色体是由人体正常的22号染色体和9号染色体相互易位形成的断裂点聚集簇-艾尔贝逊(bcr-abl)融合基因,此融合基因编码产生酪氨酸激酶活性持续激活的bcr-abl融合蛋白。目前市场上上市了针对bcr-abl为靶标的小分子酪氨酸激酶抑制剂,但都存在耐药性以及其他临床不良反应等问题。随之,研究与开发新型的bcr-abl酪氨酸激酶抑制剂已成为药学领域的热点之一。
技术实现要素:
3.本发明的目的在于提供一种噻唑并吡啶联三氮唑类化合物及其制备方法和应用。
4.为实现上述目的,本发明采用如下的技术方案:
5.一种噻唑并吡啶联三氮唑类化合物,该类化合物的结构式如下:
[0006][0007]
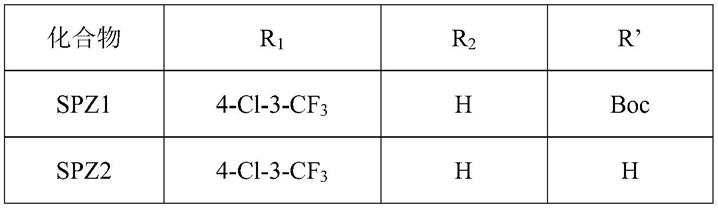
其中,r1、r2与r’具体如下:
[0008]
[0009][0010]
一种如上所述的噻唑并吡啶联三氮唑类化合物的制备方法,包括以下步骤:
[0011]
1)叔丁氧羰基保护的l-羟基脯氨酸与含取代基苯胺类化合物缩合生成氨化的boc-l-羟基脯氨酸类化合物;
[0012]
2)氨化的boc-l-羟基脯氨酸类化合物与甲基磺酰氯发生取代反应生成氨化的boc-l-甲基磺酰基脯氨酸类化合物;
[0013]
3)氮气保护下,将氨化的boc-l-甲基磺酰基脯氨酸类化合物与叠氮化钠经取代反应生成氨化的boc-l-叠氮基脯氨酸类化合物;
[0014]
4)在抗坏血酸钠和五水合硫酸铜作用下,将5-乙炔基噻唑并[5,4-b]吡啶-2-胺与氨化的boc-l-叠氮基脯氨酸类化合物缩合后得到boc保护的噻唑并吡啶联三氮唑类化合物;
[0015]
5)用多种含酰基化合物与boc保护的噻唑并吡啶联三氮唑类化合物反应,再用三氟乙酸脱去boc保护基,得到吲唑环联三氮唑类化合物。
[0016]
本发明进一步的改进在于,步骤1)中,叔丁氧羰基保护的l-羟基脯氨酸通过以下
过程制得:l-羟基脯氨酸在0℃下溶于四氢呋喃和氢氧化钠溶液中,在冰水浴下加入二碳酸二叔丁酯,然后在25~30℃下反应6h,制备得到叔丁氧羰基保护的l-羟基脯氨酸。
[0017]
本发明进一步的改进在于,步骤4)中5-乙炔基噻唑并[5,4-b]吡啶-2-胺通过以下过程制得:
[0018]
a)氮气保护下,6-溴吡啶-3-胺与硫氰化钾发生反应制备5-溴噻唑并[5,4-b]吡啶-2-胺;
[0019]
b)氮气保护下,5-溴噻唑并[5,4-b]吡啶-2-胺与三甲基硅基乙炔发生反应制备5-((三甲基甲硅烷基)乙炔基)噻唑并[5,4-b]吡啶-2-胺;
[0020]
c)用氢氧化钠的甲醇溶液将5-((三甲基甲硅烷基)乙炔基)噻唑并[5,4-b]吡啶-2-胺脱去三甲基硅基得到5-乙炔基噻唑并[5,4-b]吡啶-2-胺。
[0021]
本发明进一步的改进在于,步骤a)具体过程为:在氮气中,将kscn溶于醋酸中,冷却至0℃,加入6-溴吡啶-3-胺,然后冷却至-5℃,滴加含溴的醋酸,滴毕加热到25~30℃,搅拌,得到5-溴噻唑并[5,4-b]吡啶-2-胺;
[0022]
步骤b)具体过程为:将5-溴噻唑并[5,4-b]吡啶-2-胺、碘化铜以及四(三苯基膦)钯加入到反应容器中,然后用橡胶塞密封容器,抽真空并回填氮气,加入三乙胺后再加入三甲基硅基乙炔,加热回流反应,得到5-((三甲基甲硅烷基)乙炔基)噻唑并[5,4-b]吡啶-2-胺;
[0023]
步骤c)具体过程为:将5-((三甲基甲硅烷基)乙炔基)噻唑并[5,4-b]吡啶-2-胺溶解在二氯甲烷中,搅拌下加入氢氧化钠的甲醇溶液,然后在25~30℃下反应,得到5-乙炔基噻唑并[5,4-b]吡啶-2-胺。
[0024]
本发明进一步的改进在于,步骤1)具体过程为:将boc-l-羟脯氨酸溶于二氯甲烷中,加入三乙胺,冷却至0℃,滴加含氯甲酸乙酯的二氯甲烷溶液,反应30min~1h后,生成活性中间体;将含取代基苯胺类化合物溶液以0℃滴加到活性中间体中,在25~30℃下搅拌,得到氨化的boc-l-羟基脯氨酸类化合物;
[0025]
步骤2)具体过程为:氨化的boc-l-羟基脯氨酸类化合物溶于二氯甲烷中,冷却至0℃,加入三乙胺搅拌均匀后滴加甲基磺酰氯,在25~30℃下反应,得到氨化的boc-l-甲基磺酰基脯氨酸类化合物。
[0026]
本发明进一步的改进在于,含取代基苯胺类化合物为4-氯-3-三氟甲基苯胺,2,4-二氯苯胺,4-溴苯胺或3,4-二氟苯胺;
[0027]
步骤3)的具体过程为:氨化的boc-l-甲基磺酰基脯氨酸类化合物溶于dmf中,加入叠氮化钠,在65~70℃的氮气保护下反应,得到氨化的boc-l-叠氮基脯氨酸类化合物;
[0028]
所述步骤4)的具体过程为:将5-乙炔基噻唑并[5,4-b]吡啶-2-胺和氨化的boc-l-叠氮基脯氨酸类化合物溶于乙醇和水的混合溶剂中,然后加入抗坏血酸钠和五水合硫酸铜,在30℃下搅拌,得到boc保护基保护的噻唑并吡啶联三氮唑类化合物。
[0029]
本发明进一步的改进在于,所述步骤5)的具体过程为:boc保护基保护的吲唑联三氮唑类化合物溶于无水二氯甲烷中,加入三乙胺,在0℃下搅拌30min,滴加含有酰氯类化合物的二氯甲烷溶液,在25~30℃下反应,得到脱boc保护基的吲唑环联三氮唑类化合物。
[0030]
一种如上所述的噻唑并吡啶环联三氮唑类化合物在制备bcr-abl激酶抑制剂中的应用。
[0031]
进一步的,bcr-abl激酶为wild-abl激酶或t315i突变abl激酶。
[0032]
一种如上所述的噻唑并吡啶环联三氮唑类化合物在制备抗肿瘤药物中的应用。
[0033]
进一步的,抗肿瘤药物为抗白血病细胞的药物。
[0034]
与现有技术相比,本发明具有的有益效果:
[0035]
本发明利用圜头反应、点击化学、酰化、缩合等反应合成目标化合物,并构建了化合物库,该类化合物是具有新型分子结构的bcr-abl小分子酪氨酸激酶抑制剂,并通过ms、nmr等手段表征了目标化合物的结构。本发明基于对前期的bcr-abl酪氨酸激酶抑制剂以及bcr-abl蛋白与配体的相互作用分析等研究,采用基于片段的药物设计策略,以噻唑并吡啶联三氮唑为铰链区结合片段,l-脯氨酸为柔性linker,以构建具有激酶抑制活性的小分子化合物库,并通过adp-glo的激酶活性筛选发现具有bcr-abl激酶抑制活性的酪氨酸激酶抑制剂。激酶筛选试验表明此类化合物对abl激酶、t315i突变abl激酶均具有一定的抑制活性,其中r1为2,4-二氯,r2为环丙基甲酰基时,对abl激酶的活性最佳。细胞增殖试验表明大部分化合物对k562细胞具有一定的抑制活性。其中同样当r1为2,4-二氯,r2为环丙基甲酰基时,抗增殖活性最佳。构效关系分析发现:引入l-脯氨酸的衍生物与abl激酶的atp位点的空间匹配良好,作用模式和参照小分子伊马替尼一致。噻唑并吡啶联三氮唑对提高化合物对激酶的亲和力至关重要,同时在噻唑并吡啶环上引入酰胺侧链能够进一步提高小分子与受体的亲和力,可以作为以bcr-abl为靶标的酪氨酸激酶抑制的新型药效片段。
附图说明
[0036]
图1为本发明的合成路线图。
具体实施方式
[0037]
下面结合附图对本发明进行详细说明。
[0038]
本发明的一种噻唑并吡啶联三氮唑类化合物的结构式为:
[0039][0040]
其中,r1、r2、r’具体如下:
[0041]
[0042][0043]
参见图1,如上所述的噻唑并吡啶联三氮唑类化合物的制备方法,包括以下步骤:
[0044]
1)氮气保护下,6-溴吡啶-3-胺与硫氰化钾发生反应制备5-溴噻唑并[5,4-b]吡啶-2-胺;
[0045]
2)氮气保护下,5-溴噻唑并[5,4-b]吡啶-2-胺与三甲基硅基乙炔发生反应制备5-((三甲基甲硅烷基)乙炔基)噻唑并[5,4-b]吡啶-2-胺;
[0046]
3)用氢氧化钠的甲醇溶液将化合物5-((三甲基甲硅烷基)乙炔基)噻唑并[5,4-b]吡啶-2-胺脱去三甲基硅基得到5-乙炔基噻唑并[5,4-b]吡啶-2-胺;
[0047]
4)冰水浴中l-羟基脯氨酸与二碳酸二叔丁酯发生取代反应,制备叔丁氧羰基保护的l-羟基脯氨酸(boc-l-羟基脯氨酸);
[0048]
5)boc-l-羟基脯氨酸与含取代基苯胺类化合物缩合生成氨化的boc-l-羟基脯氨
酸类化合物;其中,含取代基苯胺类化合物为4-氯-3-三氟甲基苯胺,2,4-二氯苯胺,4-溴苯胺或3,4-二氟苯胺。
[0049]
6)氨化的boc-l-羟基脯氨酸类化合物与甲基磺酰氯发生取代反应生成氨化的boc-l-甲基磺酰基脯氨酸类化合物;
[0050]
7)氮气保护下,将氨化的boc-l-甲基磺酰基脯氨酸类化合物与叠氮化钠经取代反应生成氨化的boc-l-叠氮基脯氨酸类化合物。
[0051]
8)在抗坏血酸钠和五水合硫酸铜作用下,将5-乙炔基噻唑并[5,4-b]吡啶-2-胺与氨化的boc-l-叠氮基脯氨酸类化合物缩合后得到boc保护的噻唑并吡啶联三氮唑类化合物。
[0052]
9)用多种含酰基化合物与boc保护的噻唑并吡啶联三氮唑类化合物反应,再用三氟乙酸脱去boc保护基,得到吲唑环联三氮唑类化合物。
[0053]
所述的步骤1)具体过程为:在氮气中,将kscn溶于醋酸中,冷却0℃,加入6-溴吡啶-3-胺(10g,57.8mmol)。将混合物进一步冷却至-5℃,滴加含溴的醋酸,保持浴温低于10℃。将反应混合物加热到室温,搅拌12h。经后处理,得到黄色固体5-溴噻唑并[5,4-b]吡啶-2-胺。
[0054]
所述的步骤2)具体过程为:将5-溴噻唑并[5,4-b]吡啶-2-胺、碘化铜以及四(三苯基膦)钯加入到反应容器中。然后用橡胶塞密封容器,抽真空并回填氮气。以三乙胺为碱和溶剂,用注射器注射。室温下5min后,加入三甲基硅基乙炔,加热回流反应12h,薄层色谱法记录反应完全。经后处理纯化得到5-((三甲基甲硅烷基)乙炔基)噻唑并[5,4-b]吡啶-2-胺。
[0055]
所述的步骤3)具体过程为:将化合物5-((三甲基甲硅烷基)乙炔基)噻唑并[5,4-b]吡啶-2-胺溶解在二氯甲烷中,并在搅拌的同时缓慢加入氢氧化钠的甲醇溶液。然后混合物在室温下反应2h,然后除去有机溶剂。经后处理纯化得到5-乙炔基噻唑并[5,4-b]吡啶-2-胺。
[0056]
所述的步骤4)具体过程为:l-羟基脯氨酸在0℃下溶于四氢呋喃和氢氧化钠溶液中。在冰水浴中加入二碳酸二叔丁酯。混合物在室温下反应6h,直到薄层色谱检测到完全反应。持续冰水浴条件下,反应溶液在真空中浓缩。浓缩后的产品冰水浴中l-羟基脯氨酸与二碳酸二叔丁酯发生取代反应,制备得到叔丁氧羰基保护的l-羟基脯氨酸,无需提纯即可继续使用。
[0057]
所述的步骤5)具体过程为:将boc-l-羟脯氨酸溶于二氯甲烷中,加入三乙胺。将溶液冷却至0℃,滴加含氯甲酸乙酯的二氯甲烷溶液。反应30min~1h后,生成活性中间体。然后,将含取代基苯胺类化合物溶液以0℃滴加到上述溶液中,在室温下继续搅拌12h。经后处理得到氨化的boc-l-羟基脯氨酸类化合物。
[0058]
所述的步骤6)具体过程为:氨化的boc-l-羟基脯氨酸类化合物溶于二氯甲烷中,冷却至0℃。加入三乙胺搅拌15min。滴加甲基磺酰氯,在室温下反应12h。反应产物经后处理得到氨化的boc-l-甲基磺酰基脯氨酸类化合物。
[0059]
所述步骤7)的具体过程为:氨化的boc-l-甲基磺酰基脯氨酸类化合物溶于dmf中,加入叠氮化钠。混合物在65~70℃的氮气保护下反应16h。然后将反应溶液冷却至室温,于冰水中析出固体。后经处理纯化得到氨化的boc-l-叠氮基脯氨酸类化合物。
[0060]
所述步骤8)的具体过程为:将化合物5-乙炔基噻唑并[5,4-b]吡啶-2-胺和化合物氨化的boc-l-叠氮基脯氨酸类化合物溶于等比例的乙醇和水的混合溶剂中。然后加入抗坏血酸钠和五水合硫酸铜。悬浮液在30℃下搅拌24h,反应液在真空中浓缩,经后处理纯化后得到boc保护基保护的噻唑并吡啶联三氮唑类化合物。
[0061]
所述步骤9)的具体过程为:boc保护基保护的吲唑联三氮唑类化合物溶于无水二氯甲烷中,加入三乙胺。溶液在0℃下搅拌30min,滴加含有酰氯类化合物的二氯甲烷溶液。溶液在室温下反应2h,真空脱除二氯甲烷,用乙酸乙酯层析纯化,得到产物。之后溶于30ml无水二氯甲烷中。在0℃下,滴加三氟乙酸。溶液在室温下搅拌2h,经纯化后得到脱boc保护基的吲唑环联三氮唑类化合物。其中,酰氯类化合物为乙酰氯或环丙甲酰氯。
[0062]
一种如上所述的噻唑并吡啶联三氮唑类化合物,在制备用于抑制abl激酶、t315i突变abl激酶活性药物中的应用。
[0063]
一种如上所述的噻唑并吡啶联三氮唑类化合物在制备抗肿瘤药物中的应用。
[0064]
抗肿瘤药物为抗白血病药物。
[0065]
下面通过具体实施例进行说明。
[0066]
本发明中的室温为25-30℃,过夜为12h。
[0067]
实施例1
[0068]
一种噻唑并吡啶环联三氮唑类化合物,r1为4-cl-3-cf3,r2为h,r’为boc或h时,制备方法如下:
[0069]
1)5-溴噻唑并[5,4-b]吡啶-2-胺的合成:在氮气中,将kscn(28g,288mmol)加入500ml三颈瓶中,然后加入75ml乙酸。冷却0℃,缓慢加入6-溴吡啶-3-胺(10g,57.8mmol)。将混合物进一步冷却至-5℃,滴加4ml含有溴的醋酸,同时保持浴温低于10℃。接下来,将反应混合物加热到室温(25℃℃),搅拌一夜(12h)。将沉淀物过滤,滤液冷却至0℃,加水100ml。搅拌5min,收集形成的沉淀物,用水洗净。固体在真空下干燥过夜(12h),得到黄色固体6.8g,产率为51%。mp 218~220℃;ei-ms(m/z)231.85[m+h]
+
,229.95[m-h]-。
[0070]
2)5-((三甲基甲硅烷基)乙炔基)噻唑并[5,4-b]吡啶-2-胺的合成:将5-溴噻唑并[5,4-b]吡啶-2-胺(3.42g,16.12mmol),cui(10%,0.31g,1.61mmol),以及pd(pph3)4(10%,1.86g,1.61mmol)加入到带有冷凝管和磁力搅拌的100ml双颈圆底烧瓶中。然后用橡胶塞密封容器,抽真空并回填氮气(3次)。以三乙胺(30ml)为碱和溶剂,用注射器注射。室温下5min后,加入三甲基硅基乙炔(4.74ml,48.36mmol),加热回流反应过夜,薄层色谱法(tlc)记录反应完全。反应冷却至室温,用水(50ml)淬火。然后用乙酸乙酯(50ml)稀释溶液,过滤。用水冲洗滤液,直至水相中看不到铜配合物的蓝色。用乙酸乙酯(30ml
×
3)提取复合水层。结合有机层在无水硫酸钠上干燥,过滤,蒸发溶剂。粗品经快速柱层析(体积比石油醚:乙酸乙酯=5:1)纯化得到产品,产率为72%。ei-ms(m/z)248.05[m+h]
+
,246.30[m-h]-。
[0071]
3)5-乙炔基噻唑并[5,4-b]吡啶-2-胺的合成:将化合物5-((三甲基甲硅烷基)乙炔基)噻唑并[5,4-b]吡啶-2-胺(2.77g,13.16mmol)溶解在30ml二氯甲烷中,并在搅拌的同时缓慢加入30ml naoh的甲醇溶液(质量浓度3%)。然后混合物在室温下反应2h,然后除去有机溶剂。通过色谱法(体积比pe:etoac=1:1)进一步纯化残余物以产生呈黄色固体状的化合物1.98g,产率86%。mp 225~227℃;ei-ms(m/z)176.15[m+h]
+
,174.20[m-h]-。
[0072]
4)boc-l-羟基脯氨酸的制备:l-羟基脯氨酸(9.0g,68.7mmol)在0℃下溶于四氢呋
(4-氯-3-(三氟甲基)苯基)吡咯烷-2-甲酰胺(spz2)的制备:化合物(2s,4s)-4-(4-(2-氨基噻唑并[5,4-b]吡啶-5-基)-1h-1,2,3-三氮唑-1-基)-2-((4-氯-3-(三氟甲基)苯基)氨基甲酰基)吡咯烷-1-羧酸叔丁酯溶于30ml无水二氯甲烷中。在0℃下,滴加2ml三氟乙酸。溶液在室温下搅拌2h,用50ml二氯甲烷稀释,用碳酸氢钠溶液调节ph值至8,用水洗涤3次,每次30ml。有机层用无水硫酸钠干燥,过滤,经色谱层析纯化(体积比石油醚:乙酸乙酯=1;3),得到产物(spz2),产率为36%。mp 123~125℃;ei-ms(m/z)509.15[m+h]
+
,507.15[m-h]-。hrms m/z对c
20h17
clf3n8os([m+h]
+
)计算值为509.08867,实测值为509.09129。1h nmr(400mhz,dmso-d6)δ10.40(s,1h),8.65(s,1h),8.23(s,1h),7.89
–
7.80(m,3h),7.74
–
7.71(m,1h),7.68
–
7.66(m,1h),7.65
–
7.59(m,1h),5.23
–
5.14(m,1h),4.03
–
3.93(m,1h),3.53
–
3.45(m,2h),2.91
–
2.73(m,1h),2.48
–
2.43(m,1h),1.91(s,1h)。
[0078]
实施例2
[0079]
一种噻唑并吡啶环联三氮唑类化合物,r1为4-cl-3-cf3,r2为乙酰基,r’为h时,制备方法如下:
[0080]
步骤1)~步骤8)同实施例1,得到(2s,4s)-4-(4-(2-氨基噻唑并[5,4-b]吡啶-5-基)-1h-1,2,3-三氮唑-1-基)-2-((4-氯-3-(三氟甲基)苯基)氨基甲酰基)吡咯烷-1-羧酸叔丁酯。
[0081]
9)(2s,4s)-4-(4-(2-乙酰氨基噻唑并[5,4-b]吡啶-6-基)-1h-1,2,3-三氮唑-1-基)-n-(4-氯-3-(三氟甲基)苯基)吡咯烷-2-甲酰胺(spz3)的制备:化合物(2s,4s)-4-(4-(2-氨基噻唑并[5,4-b]吡啶-5-基)-1h-1,2,3-三氮唑-1-基)-2-((4-氯-3-(三氟甲基)苯基)氨基甲酰基)吡咯烷-1-羧酸叔丁酯溶于30ml无水二氯甲烷中,加入三乙胺(0.37ml,2.7mmol)。溶液在0℃下搅拌30min,滴加含有乙酰氯的二氯甲烷(2ml)。溶液在室温下反应2h,真空脱除二氯甲烷,用色谱层析纯化,得到产物(2s,4s)-4-(4-(2-乙酰氨基噻唑并[5,4-b]吡啶-6-基)-1h-1,2,3-三氮唑-1-基)-2-((4-氯-3-(三氟甲基)苯基)氨基甲酰基)吡咯烷-1-羧酸叔丁酯。之后产物溶于30ml无水二氯甲烷中。在0℃下,滴加2ml三氟乙酸。溶液在室温下搅拌2h,用50ml二氯甲烷稀释,用碳酸氢钠溶液调节ph值至8,用水洗涤3次,每次30ml。有机层用无水硫酸钠干燥,过滤,经色谱层析纯化(体积比石油醚:乙酸乙酯=1:3)得到化合物(spz3),产率为30%。mp 137~139℃;ei-ms(m/z)551.15[m+h]
+
。hrms m/z对c
22h19
clf3n8o2s([m+h]
+
)计算值为551.09923,实测值为551.09943。1h nmr(400mhz,dmso-d6)δ12.52(s,1h),10.39(s,1h),8.81(s,1h),8.20(d,j=2.4hz,1h),8.14(d,j=8.5hz,1h),8.05(d,j=8.5hz,1h),7.93(dd,j=8.8,2.2hz,1h),7.59(d,j=8.8hz,1h),5.24
–
5.17(m,1h),4.01
–
3.97(m,1h),3.53
–
3.48(m,1h),3.43-3.41(m,1h),2.87
–
2.77(m,1h),2.55-2.53(m,1h),2.23(s,4h)。
[0082]
实施例3
[0083]
一种噻唑并吡啶环联三氮唑类化合物,r1为4-cl-3-cf3,r2为环丙甲酰基,r’为h时,制备方法同实施例2,将实施例2中的乙酰氯替换为环丙甲酰氯,制备得到(2s,4s)-n-(4-氯-3-(三氟甲基)苯基)-4-(4-(2-(环丙烷甲酰胺基)噻唑并[5,4-b]吡啶-6-基)-1h-1,2,3-三氮唑-1-基)吡咯烷-2-甲酰胺(spz4),经色谱纯化(体积比石油醚:乙酸乙酯=1:2)后得到产物,产率为41%。mp 159~161℃;ei-ms(m/z)577.15[m+h]
+
,575.20[m-h]-。hrms m/z对c
24h21
clf3n8o2s([m+h]
+
)计算值为577.11488,实测值为577.11560。1h nmr(400mhz,
dmso-d6)δ12.84(s,1h),10.37(s,1h),8.82(s,1h),8.21(s,1h),8.14(d,j=8.4hz,1h),8.05(d,j=8.5hz,1h),7.93(d,j=9.0hz,1h),7.59(d,j=8.7hz,1h),5.23
–
5.14(m,1h),3.99
–
3.95(m,1h),3.51
–
3.47(m,2h),2.85
–
2.78(m,1h),2.47
–
2.45(m,1h),2.05
–
2.01(m,1h),1.91(s,1h),1.00(d,j=7.9hz,4h)。
[0084]
实施例4
[0085]
一种噻唑并吡啶环联三氮唑类化合物,r1为2,4-di-cl,r2为h,r’为boc或h时,制备方法如同实施例1,将实施例1中的5-氨基-2-氯三氟甲基苯替换为2,4-二氯苯胺,制备得到(2s,4s)-4-(4-(2-氨基噻唑并[5,4-b]吡啶-5-基)-1h-1,2,3-三唑-1-基)-2-((2,4-二氯苯基)氨基甲酰基)吡咯烷-1-羧酸叔丁酯(spz5)和(2s,4s)-4-(4-(2-氨基噻唑并[5,4-b]吡啶-5-基)-1h-1,2,3-三唑-1-基)-n-(2,4-二氯苯基)吡咯烷-2-甲酰胺(spz6)。
[0086]
化合物spz5经乙酸乙酯色谱纯化得到固体化合物0.80g,产率为66%。mp 134~136℃;ei-ms(m/z)573.25[m-h]-.hrms m/z对c
24h25
cl2n8o3s([m+h]
+
)计算值为575.11474,实测值为575.11529.1h nmr(400mhz,dmso-d6)δ9.72(s,1h),8.70(s,1h),8.18
–
8.09(m,1h),7.88(s,2h),7.71
–
7.66(m,3h),7.37(s,1h),5.37
–
5.31(m,1h),4.59
–
4.56(m,1h),4.15
–
4.12(m,1h),3.87
–
3.84(m,1h),3.06
–
2.93(m,1h),2.76
–
2.64(m,1h),1.46(s,3h),1.36(s,6h)。
[0087]
化合物spz6经色谱纯化(体积比石油醚:乙酸乙酯=1:5)得到固体化合物,产率为51%。mp 200~202℃;ei-ms(m/z)475.15[m+h]
+
,473.05[m-h]-。hrms m/z对c
19h17
cl2n8os([m+h]
+
)计算值为475.06231,实测值为475.06292。1h nmr(400mhz,dmso-d6)δ10.33(s,1h),8.63(s,1h),8.26(d,j=8.9hz,1h),7.85(s,2h),7.79(d,j=8.3hz,1h),7.65(d,j=8.3hz,1h),7.59(d,j=2.4hz,1h),7.37(dd,j=8.9,2.4hz,1h),5.18
–
5.10(m,1h),4.06
–
3.99(m,1h),3.57
–
3.52(m,1h),3.20
–
3.12(m,1h),2.87
–
2.81(m,1h),2.55
–
2.52(m,1h),1.88(s,1h)。
[0088]
实施例5
[0089]
一种噻唑并吡啶环联三氮唑类化合物,r1为2,4-di-cl,r2为乙酰基,r’为h时,制备方法如同实施例2。将实施例2中的5-氨基-2-氯三氟甲基苯替换为2,4-二氯苯胺,制备得到(2s,4s)-4-(4-(2-乙酰氨基噻唑并[5,4-b]吡啶-5-基)-1h-1,2,3-三氮唑-1-基)-n-(2,4-二氯苯基)吡咯烷-2-甲酰胺(spz7)。经色谱层析纯化(体积比乙酸乙酯:甲醇=1:1)后得到产物,产率为84%。mp 198~200℃;ei-ms(m/z)517.20[m+h]
+
,515.25[m-h]-。hrms m/z对c
21h19
cl2n8o2s([m+h]
+
)计算值为517.07287,实测值为517.07356。1h nmr(400mhz,dmso-d6)δ12.53(s,1h),10.34(s,1h),8.79(s,1h),8.22(d,j=8.6hz,1h),8.13(d,j=8.5hz,1h),8.02(d,j=8.5hz,1h),7.56(d,j=2.3hz,1h),7.35(dd,j=8.9,2.3hz,1h),5.22
–
5.16(m,1h),4.14
–
4.02(m,1h),3.60
–
3.56(m,1h),3.48
–
3.46(m,1h),2.93
–
2.82(m,1h),2.63
–
2.55(m,1h),2.23(s,3h),1.91(s,1h)。
[0090]
实施例6
[0091]
一种噻唑并吡啶环联三氮唑类化合物,r1为2,4-di-cl,r2为环丙甲酰基,r’为h时,制备方法如同实施例3。将实施例3中的5-氨基-2-氯三氟甲基苯替换为2,4-二氯苯胺,制备得到(2s,4s)-4-(4-(2-(环丙烷甲酰胺基)噻唑并[5,4-b]吡啶-6-基)-1h-1,2,3-三唑-1-基)-n-(2,4-二氯苯基)吡咯烷-2-甲酰胺(spz8)。经色谱层析纯化(体积比乙酸乙酯:甲醇
=1:1)后得到产物,产率为55%。mp 202~204℃;ei-ms(m/z)543.20[m+h]
+
,541.25[m-h]-。hrms m/z对c
23h21
cl2n8o2s([m+h]
+
)计算值为543.08852,实测值为543.08965。1h nmr(400mhz,dmso-d6)δ12.84(s,1h),10.33(s,1h),8.79(s,1h),8.26(d,j=8.9hz,1h),8.13(d,j=8.5hz,1h),8.01(d,j=8.5hz,1h),7.54(d,j=2.4hz,1h),7.34(dd,j=8.9,2.4hz,1h),5.19
–
5.13(m,1h),4.08
–
3.99(m,1h),3.62
–
3.51(m,2h),2.91
–
2.80(m,1h),2.60
–
2.55(m,1h),2.07
–
2.00(m,1h),1.91(s,1h),1.01
–
0.98(m,4h)。
[0092]
实施例7
[0093]
一种噻唑并吡啶环联三氮唑类化合物,r1为4-br,r2为h,r’为boc或h时,制备方法如同实施例1。将实施例1中的5-氨基-2-氯三氟甲基苯替换为4-溴苯胺,制备得到(2s,4s)-4-(4-(2-氨基噻唑并[5,4-b]吡啶-5-基)-1h-1,2,3-三唑-1-基)-2-((4-溴苯基)氨基甲酰基)吡咯烷-1-羧酸叔丁酯(spz9)和(2s,4s)-4-(4-(2-氨基噻唑并[5,4-b]吡啶-5-基)-1h-1,2,3-三唑-1-基)-n-(4-溴苯基)吡咯烷-2-甲酰胺(spz10)。
[0094]
化合物spz9经乙酸乙酯色谱纯化得到产物0.60g,产率为67%。mp 176~178℃;ei-ms(m/z)583.25[m-h]-.hrms m/z对c
24h26
brn8o3s([m+h]
+
)计算值为585.10320,实测值为585.10303.1h nmr(400mhz,dmso-d6)δ10.24(s,1h),8.71(s,1h),7.88(s,3h),7.69(d,j=8.4hz,1h),7.59-7.55(m,2h),7.49(d,j=8.5hz,2h),5.37-5.30(m,1h),4.44
–
4.38(m,1h),4.19
–
4.10(m,1h),3.83
–
3.72(m,1h),3.04
–
2.92(m,1h),2.62
–
2.55(m,1h),1.43(s,3h),1.29(s,6h)。
[0095]
化合物spz10经色谱纯化(体积比石油醚:乙酸乙酯=1:3)后得到固体化合物,产率为60%。mp 152~154℃;ei-ms(m/z)485.10[m+h]
+
,483.10[m-h]-。hrms m/z对c
19h18
brn8os([m+h]
+
)计算值为485.05077,实测值为485.05200。1h nmr(400mhz,dmso-d6)δ10.11(s,1h),8.66(s,1h),7.91
–
7.83(m,3h),7.68(d,j=8.3hz,1h),7.62(d,j=8.8hz,2h),7.45(d,j=8.8hz,2h),5.21
–
5.15(m,1h),3.95
–
3.91(m,1h),3.48
–
3.43(m,1h),3.35
–
3.29(m,1h),2.83
–
2.77(m,1h),2.42
–
2.33(m,1h),1.90(s,1h)。
[0096]
实施例8
[0097]
一种噻唑并吡啶环联三氮唑类化合物,r1为4-br,r2为乙酰基,r’为h时,制备方法如同实施例2。将实施例2中的5-氨基-2-氯三氟甲基苯替换为4-溴苯胺,制备得到(2s,4s)-4-(4-(2-乙酰氨基噻唑并[5,4-b]吡啶-6-基)-1h-1,2,3-三氮唑-1-基)-n-(4-溴苯基)吡咯烷-2-甲酰胺(spz11)。经色谱层析纯化(体积比石油醚:乙酸乙酯=1:2)后得到产物,产率为39%。mp 173~175℃;ei-ms(m/z)526.95[m+h]
+
,525.10[m-h]-.hrms m/z对c
21h20
brn8o2s([m+h]
+
)计算值为527.06133,实测值为527.06202。1h nmr(400mhz,dmso-d6)δ12.54(s,1h),10.12(s,1h),8.82(s,1h),8.16(d,j=8.4hz,1h),8.08(d,j=8.4hz,1h),7.62(d,j=8.8hz,2h),7.44(d,j=8.8hz,2h),5.26
–
5.18(m,1h),3.97
–
3.93(m,1h),3.49
–
3.46(m,2h),2.87
–
2.79(m,1h),2.47
–
2.39(m,1h),2.23(s,3h),1.91(s,1h)。
[0098]
实施例9
[0099]
一种噻唑并吡啶环联三氮唑类化合物,r1为4-br,r2为环丙甲酰基时,制备方法如同实施例3。将实施例3中的5-氨基-2-氯三氟甲基苯替换为4-溴苯胺,制备得到(2s,4s)-n-(4-溴苯基)-4-(4-(2-(环丙烷甲酰胺基)噻唑并[5,4-b]吡啶-6-基)-1h-1,2,3-三唑-1-基)吡咯烷-2-甲酰胺(spz12)。经色谱层析纯化(体积比石油醚:乙酸乙酯=1:3)后得到固
体产物,产率为43%。mp 156~158℃;ei-ms(m/z)553.10[m+h]
+
,551.15[m-h]-。hrms m/z对c
23h22
brn8o2s([m+h]
+
)计算值为553.07698,实测值为553.07346.1h nmr(400mhz,dmso-d6)δ12.86(s,1h),10.11(s,1h),8.83(s,1h),8.16(d,j=8.4hz,1h),8.08(d,j=8.4hz,1h),7.61(d,j=8.8hz,2h),7.44(d,j=8.8hz,2h),5.23
–
5.17(m,1h),3.96
–
3.92(m,1h),3.48
–
3.44(m,2h),2.86
–
2.79(m,1h),2.46
–
2.38(m,1h),2.07
–
2.01(m,1h),1.91(s,1h),1.01
–
0.98(m,4h)。
[0100]
实施例10
[0101]
一种噻唑并吡啶环联三氮唑类化合物,r1为3,4-di-f,r2为h时,r’为boc或h时,制备方法如同实施例1。将实施例1中的5-氨基-2-氯三氟甲基苯替换为3,4-二氟苯胺,制备得到(2s,4s)-4-(4-(2-氨基噻唑并[5,4-b]吡啶-5-基)-1h-1,2,3-三唑-1-基)-2-((3,4-二氟苯基)氨基甲酰基)吡咯烷-1-羧酸叔丁酯(spz13)和(2s,4s)-4-(4-(2-氨基噻唑并[5,4-b]吡啶-5-基)-1h-1,2,3-三唑-1-基)-n-(3,4-二氟苯基)吡咯烷-2-甲酰胺(spz14)。
[0102]
化合物spz13经乙酸乙酯纯化得到产物0.70g,产率为84%。mp 137~139℃;ei-ms(m/z)541.30[m-h]-.hrms m/z对c
24h25
f2n8o3s([m+h]
+
)计算值为543.17384,实测值为543.17396.1h nmr(400mhz,dmso-d6)δ10.13(s,1h),8.69(s,1h),7.92(d,j=5.0hz,1h),7.71(d,j=5.8hz,2h),7.59
–
7.54(m,1h),7.51(d,j=8.3hz,1h),7.20
–
7.13(m,1h),7.12
–
7.08(m,1h),5.24
–
5.11(m,1h),4.30
–
4.17(m,1h),4.01
–
3.94(m,1h),3.67
–
3.59(m,1h),2.86
–
2.75(m,1h),2.47
–
2.33(m,1h),1.25(s,3h),1.12(s,6h)。
[0103]
化合物spz14经色谱纯化(体积比石油醚:乙酸乙酯=1:1)后得到固体化合物,产率为74%。mp 155~157℃;ei-ms(m/z)443.05[m+h]
+
,441.20[m-h]-.hrms m/z对c
19h17
f2n8os([m+h]
+
)计算值为443.12141,实测值为443.12341。1h nmr(400mhz,dmso-d6)δ10.19(s,1h),8.64(s,1h),7.88
–
7.83(m,3h),7.83
–
7.77(m,1h),7.67(d,j=8.3hz,1h),7.42(d,j=9.0hz,1h),7.37
–
7.31(m,1h),5.24
–
5.13(m,1h),3.95
–
3.92(m,1h),3.52
–
3.44(m,1h),3.33
–
3.29(m,1h),2.85
–
2.77(m,1h),2.44
–
2.37(m,1h),1.90(s,1h)。
[0104]
实施例11
[0105]
一种噻唑并吡啶环联三氮唑类化合物,r1为3,4-di-f,r2为乙酰基,r’为h时,制备方法如同实施例2。将实施例2中的5-氨基-2-氯三氟甲基苯替换为3,4-二氟苯胺,制备得到(2s,4s)-4-(4-(2-乙酰氨基噻唑并[5,4-b]吡啶-5-基)-1h-1,2,3-三氮唑-1-基)-n-(3,4-二氟苯基)吡咯烷-2-甲酰胺(spz15)。经色谱层析纯化(体积比石油醚:乙酸乙酯=1:2)后得到产物,产率为50%。mp 129~131℃;ei-ms(m/z)485.25[m+h]
+
,483.30[m-h]-。hrms m/z对c
21h19
f2n8o2s([m+h]
+
)计算值为485.13197,实测值为485.12879.1h nmr(400mhz,dmso-d6)δ12.54(s,1h),10.20(s,1h),8.81(s,1h),8.16(d,j=8.5hz,1h),8.08(d,j=8.7hz,1h),7.82
–
7.77(m,1h),7.43
–
7.38(m,1h),7.37-7.35(m,1h),5.25
–
5.16(m,1h),3.97-3.93(m,1h),3.51
–
3.47(m,2h),2.86
–
2.79(m,1h),2.51
–
2.42(m,1h),2.23(s,3h),1.91(s,1h)。
[0106]
实施例12
[0107]
一种噻唑并吡啶环联三氮唑类化合物,r1为3,4-di-f,r2为环丙甲酰基,r’为h时,制备方法如同实施例3。将实施例3中的5-氨基-2-氯三氟甲基苯替换为3,4-二氟苯胺,制备得到(2s,4s)-4-(4-(2-(环丙烷甲酰胺基)噻唑并[5,4-b]吡啶-5-基)-1h-1,2,3-三唑-1-基)-n-(3,4-二氟苯基)吡咯烷-2-甲酰胺(spz16)。经色谱层析纯化(体积比石油醚:乙酸乙
酯=1:2)后得到固体产物,产率为69%。mp 133~135℃;ei-ms(m/z)511.20[m+h]
+
,509.30[m-h]-。hrms m/z对c
23h21
f2n8o2s([m+h]
+
)计算值为511.14762,实测值为511.14361.1h nmr(400mhz,dmso-d6)δ12.87(s,1h),10.46(s,1h),8.88(s,1h),8.18(d,j=8.5hz,1h),8.10(d,j=8.5hz,1h),7.77
–
7.72(m,1h),7.40
–
7.38(m,1h),7.37-7.35(m,1h),5.39
–
5.33(m,1h),4.28
–
4.20(m,1h),3.71
–
3.58(m,2h),3.01
–
2.93(m,1h),2.65
–
2.59(m,1h),2.06
–
2.00(m,1h),1.91(s,1h),1.03
–
0.95(m,4h)。
[0108]
下面对本发明制得的具有抗肿瘤活性的噻唑并吡啶联三氮唑类化合物进行bcr-abl激酶抑制活性筛选。
[0109]
测定方法具体如下:
[0110]
激酶bcr-abl、bcr-abl(t315i)和底物abltide购自signal-chem公司,选用promega公司的adp-glo
tm kinase assays检测试剂盒检测目标化合物的抑酶活性,操作方法按照试剂盒说明进行。
[0111]
abl实验中,用buffer(2
×
)(tris 80mm,mgcl
2 20mm,bsa 0.2mg/ml,dtt 2mm)将atp(1mm)稀释到80倍并配制成atp(125μm)的buffer(2
×
)溶液;再将atp(125μm)溶液和abltide溶液按体积1:1配制成atp(62.5μm)-abltide(0.5μg/μl)的混合溶液备用;用buffer(1
×
)(tris 40mm,mgcl
2 10mm,bsa 0.1mg/ml,dtt 1mm)将abl激酶溶液稀释到100倍并配制成abl(1ng/μl)的buffer(1
×
)溶液备用。
[0112]
abl(t315i)实验中,atp-abltide以及abl(t315i)的配制步骤同上,不同的是此实验中,atp和abl(t315i)的浓度分别为12.5μm和2ng/μl。
[0113]
用buffer(1
×
)将四个目标化合物分别配制成1.5
×
10-5
,1.5
×
10-6
,1.5
×
10-7
,1.5
×
10-8
,1.5
×
10-9
,1.5
×
10-10
mol/l浓度梯度的样品溶液,于384孔板上每孔依次加入2μl atp-abltide的混合溶液,1μl样品溶液,2μl酶溶液;空白孔加3μl缓冲液和2μl atp-abltide的混合溶液;对照孔每孔加2μl atp-abltide的混合溶液,1μl缓冲液,2μl酶溶液,加毕,30℃下孵育60min;加入adp-glo试剂5μl,在25℃下孵育40min;最后加入kinase detection试剂,再在25℃下孵育30min。采用perkinelmer多功能酶标仪的化学发光模块测定每孔的发光值,计算化合物对abl的抑制率和ic
50
。
[0114]
本发明的一种噻唑并吡啶联三氮唑类化合物的结构式为:
[0115][0116]
上述结构式的噻唑并吡啶联三氮唑类化合物的激酶抑制活性如表1所示
[0117]
表1噻唑并吡啶联三氮唑类化合物对bcr-abl
wt
和bcr-abl
t315i
抑制活性ic
50
(nm)
[0118]
[0119][0120]
化合物spz1-spz16对bcr-abl
wt
和bcr-abl
t315i
的激酶抑制活性的测定结果汇总在表1。可以看出,当噻唑并[5,4-b]吡啶作为核心结构与不同结合基团相互作用时,大部分的化合物对bcr-abl激酶具有较好的抑制活性,其中活性最好的是化合物spz2、spz8和spz12其ic
50
值分别为1.60nm、0.60nm和3.58nm。而在bcr-abl
t315i
激酶,化合物spz2、spz3、spz9和spz12表现出了较好的活性,其ic
50
值为9.69nm、59.62nm、25.21nm和49.95nm。由上表中的数据可以发现,氨基乙酰化有利于噻唑并[5,4-b]吡啶衍生物对bcr-abl激酶的活性(spz3,spz4,spz7,spz8,spz11,spz12,spz15,spz16》spz2,spz6,spz10,spz14)。一般来说,环丙基甲酰化比乙酰化的氨基更好,可以提高该系列的活性。同时,卤化取代基也被引入到端部苯环上,这些取代基也影响了对激酶的活性,不同取代基与抑制活性之间的关系可以大致概括为:2,4-二氯取代基》4-溴取代基》4-氯-3-三氟甲基取代基》3,4-二氟取代基。活性结果表明,取代基的不同会直接影响到化合物对激酶的抑制活性。
[0121]
下面测定噻唑并吡啶联三氮唑类化合物对肿瘤细胞的增值抑制活性。
[0122]
采用mtt法测定噻唑并吡啶联三氮唑类化合物对肿瘤细胞的增值抑制活性作用。
[0123]
本发明提供的噻唑并吡啶联三氮唑类化合物具有抗肿瘤的作用。对肿瘤细胞具有体外抑制增值活性效果,在人白血病细胞(k562细胞)中具有抑制肿瘤细胞的增值活性效果,可用于对白血病的治疗。
[0124]
将处于生长指数期的人白血病细胞(k562细胞)用rpmi1640培养基稀释成104个/
ml数量级的细胞溶液,平行接种于96孔培养板中(2000-4000个/孔),每孔接种体积为180μl,并在37℃、5%co2下培养12h;
[0125]
每孔加入不同浓度的待测化合物20μl,得到孔中化合物的最终浓度为:1.5
×
10-7
mol/l,1.5
×
10-6
mol/l,1.5
×
10-5
mol/l,1.5
×
10-4
mol/l,每个浓度设3个复孔,阴性对照设6个复孔,每个孔加细胞不加化合物,尼罗替尼或伊马替尼为阳性对照,继续在37℃、5%co2下培养48h;
[0126]
每孔加入mtt(5mg/ml)20μl,得到每个孔中mtt的终浓度为0.5mg/ml,在37℃、5%co2下培养4h,小心吸去上清,每孔再加dmso 150μl,振荡15min,用酶联免疫检测仪测量各孔490nm处的紫外吸收值(od值),然后计算细胞抑制率,并根据抑制率采用线性回归法计算求出化合物的ic
50
值;
[0127]
细胞抑制率的计算公式为:
[0128]
抑制率%=(对照孔平均od值-用药组平均od值)/对照孔平均od值
×
100%;
[0129]
检测结果显示:与阴性对照组相比,噻唑并吡啶联三氮唑类化合物对上述肿瘤细胞具有不同程度的体外抑制作用,如表2所示。
[0130]
k562细胞增殖活性:
[0131]
表2含有噻唑并吡啶联三氮唑类化合物对k562细胞抑制活性ic
50
(μm)
[0132][0133]
细胞活性筛选试验表明了化合物spz1-spz16对k562细胞都具有一定的细胞增值抑制活性,其ic
50
值在都微摩尔水平,范围为0.74μm-62.96μm。其中活性最好的化合物是spz8,ic
50
值为0.74μm。对噻唑并[5,4-b]吡啶衍生物来讲,在端部苯环上引入了卤化取代基时,这些取代基对肿瘤细胞的抗增殖活性有明显的影响,其中取代基为2,4-二氯的化合物活性最好。另外,化合物spz2、spz12和spz13对k562细胞也具有较好的细胞抑制活性,可以展开进一步的深入研究。
[0134]
本发明采用基于片段的药物设计策略,在对噻唑并吡啶环上氨基酰化的同时,在端部苯环上引入了卤化取代基,以构建具有激酶抑制活性的噻唑并吡啶联三氮唑类化合物库,并通过激酶活性筛选发现具有bcr-abl激酶抑制活性的酪氨酸激酶抑制剂spz2、spz8、spz12和spz13。这四个化合物能够用于制备抗肿瘤(慢性粒细胞白血病)药物中,具有一定的抑制bcr-abl、bcr-abl
t315i
激酶活性,并且对k562细胞具有一定的细胞增值抑制活性。端部苯环上引入的卤化取代基,可以扩展bcr-abl激酶抑制剂的结构多样性,同时活性试验显示噻唑并吡啶环对化合物的激酶抑制活性具有重要作用,能够提高受体与化合物之间的亲和力,可以作为bcr-abl酪氨酸激酶抑制剂的药效片段。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1