醌式喹啉类化合物及其制备方法和用途
1.本发明涉及一种醌式喹啉类化合物及其制备方法和用途。
背景技术:
2.山沉香是木犀科(oleaceae)丁香属植物羽叶丁香syringa pinnatifolia hemsl.的去皮的根、茎及粗枝,集中分布于内蒙古阿拉善与宁夏的贺兰山地区。山沉香中主要含有木脂素与倍半萜类成分,常用于治疗心肌缺血、胸闷气短等心肺疾病。
3.cn104606176a公开了一种由山沉香提取得到的单环半倍萜类化合物,该化合物能够用于治疗癌症。cn105982966a公开了一种山沉香提取物,该提取物由山沉香经有机溶剂超声提取,提取液浓缩后,用硅胶柱分离得到。所得提取物能够用于制备抗肿瘤药物。cn109020986a公开了一种喹啉醌并杂环衍生物,其能够用于制备抗肿瘤药物。cn109121411a公开了一种嘧啶-异喹啉-醌衍生物,其能够治疗细菌性疾病和抗性细菌性疾病。
技术实现要素:
4.本发明的一个目的在于提供一种醌式喹啉类化合物,其具有抗肿瘤和抗菌活性。本发明的另一个目的在于提供上述化合物的制备方法。本发明再一个目的在于提供上述化合物的用途。
5.一方面,本发明提供了一种醌式喹啉类化合物具有式(i)所示的结构或其异构体或其药学上可接受的盐:
[0006][0007]
其中,r1和r4分别独立地选自氢、卤素、c1-c6烷基、c3-c6环烷基、c2-c6杂烷基、c1-c6烷氧基或c1-c6烷硫基;
[0008]
r2选自氢、卤素、c1-c6烷基、c3-c6环烷基、c2-c6杂烷基、c1-c6烷氧基、c1-c6烷硫基或其中,r6选自c1-c6亚烷基、c2-c6杂亚烷基,r5选自氢、卤素、c1-c6烷基、c2-c6杂烷基;
[0009]
r3选自氢、c1-c6烷基、c3-c6环烷基、c2-c6杂烷基、c1-c6烷氧基、c1-c6烷硫基或
c1-c6酯基。
[0010]
根据本发明的醌式喹啉类化合物,优选地,杂烷基或杂亚烷基中的杂原子选自n、o、s中的一种或多种。
[0011]
根据本发明的醌式喹啉类化合物,优选地,r1为c1-c6烷氧基;
[0012]
r4选自氢或c1-c6烷基;r3为c1-c6酯基;r2为其中,r6选自c1-c6亚烷基,r5选自c1-c6烷基。
[0013]
根据本发明的醌式喹啉类化合物,优选地,r1为c1-c3烷氧基;r4为氢;r3为c1-c3酯基;r2为其中,r6选自c1-c3亚烷基,r5选自c1-c3烷基。
[0014]
根据本发明的醌式喹啉类化合物,优选地,所述醌式喹啉类化合物选自如下所示的化合物或其异构体或其药学上可接受的盐:
[0015][0016]
根据本发明的醌式喹啉类化合物,优选地,所述醌式喹啉类化合物选自如下所示的化合物或其药学上可接受的盐:
[0017][0018]
根据本发明的醌式喹啉类化合物,优选地,药学上可接受的盐选自盐酸盐、苯磺酸盐、甲苯磺酸盐、甲磺酸盐、酒石酸盐、苹果酸盐、马来酸盐、富马酸盐、氢溴酸盐、硫酸盐、乙磺酸盐中的一种或多种。
[0019]
另一方面,本发明提供了上述醌式喹啉类化合物的制备方法,包括如下步骤:
[0020]
(1)将山沉香醇提浸膏采用正相硅胶色谱柱,以石油醚-乙酸乙酯-甲醇系统,二氯甲烷-乙酸乙酯-甲醇系统,二氯甲烷-甲醇系统作为溶剂洗脱,得到第一流分;
[0021]
(2)将第一流分采用葡聚糖凝胶色谱柱,以二氯甲烷-甲醇系统作为溶剂洗脱,得到第二流分;
[0022]
(3)将第二流分采用ods色谱柱,以甲醇-水系统作为溶剂洗脱,得到第三流分;
[0023]
(4)将第三流分采用葡聚糖凝胶色谱柱,以甲醇作为溶剂洗脱,得到第四流分;
[0024]
(5)将第四流分采用半制备液相色谱仪,以甲醇水溶液作为流动相分离,得到醌式喹啉类化合物。
[0025]
再一方面,本发明提供了上述醌式喹啉类化合物在制备抗肿瘤和/或抗菌药物中的用途。
[0026]
根据本发明的用途,优选地,所述肿瘤选自肝癌、乳腺癌、前列腺癌、皮肤癌、头颈癌、肺癌、食道癌、宫颈癌、胰腺癌、结肠癌、肾癌、输尿管癌、膀胱癌中的一种或多种;所述菌选自枯草芽孢杆菌、白色念珠菌、金黄色葡萄球菌、铜绿假单胞菌、沙门菌中的一种或多种。
[0027]
本发明意外地发现,本发明醌式喹啉类化合物具有优异的抗肿瘤和抗菌活性。
附图说明
[0028]
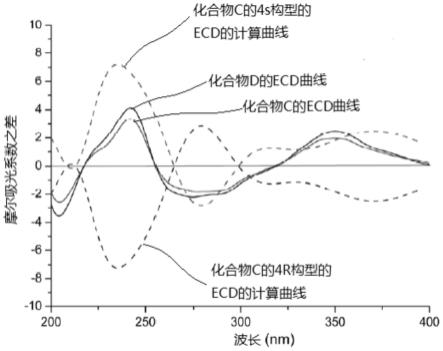
图1为ecd图谱。
[0029]
图2为化合物c的浓度与hepg2细胞存活率的关系图。
[0030]
图3为化合物c的浓度与mcf-7细胞存活率的关系图。
[0031]
图4为化合物d的浓度与hepg2细胞存活率的关系图。
[0032]
图5为化合物d的浓度与mcf-7细胞存活率的关系图。
[0033]
图6为化合物d的浓度与pc-3m-ie8细胞存活率的关系图。
[0034]
图2~6中,**表示p《0.01,***表示p《0.001。
具体实施方式
[0035]
下面结合具体实施例对本发明作进一步的说明,但本发明的保护范围并不限于此。
[0036]
《术语解释》
[0037]
在本发明中,cm-cn表示具有m~n个碳原子;举例来说,c1-c10烷基表示具有1~10个碳原子的烷基。
[0038]
在本发明中,“烷基”表示具有一个连接点的、衍生自直链的或支链的脂族烃的基团。“杂烷基”表示具有一个连接点的、具有至少一个杂原子的烷基。“环烷基”表示具有一个连接点的、衍生自脂族环烃的基团。前缀“杂”表示一个或多个碳原子已被不同的原子置换。
[0039]
除非特别声明,所有基团可为取代或未取代的。根据本发明的一些具体实施方式,取代基选自卤素、烷基、烷氧基、芳基。
[0040]
除非另有定义,本文所用的所有技术和科学术语的含义均与本发明所属领域的普通技术人员通常理解的一样。尽管与本文所述的方法和材料类似或等同的方法和材料也可用于本发明的实施或测试中,但是下文描述了合适的方法和材料。所有的出版物、专利申请、专利、以及本文提及的其它参考资料均以引用方式全文并入本文。如发生矛盾,以本说明书及其包括的定义为准。此外,材料、方法、制备例和实施例仅是示例性的,并不旨在进行限制。
[0041]
《醌式喹啉类化合物》
[0042]
本发明的醌式喹啉类化合物具有式(i)所示的结构或其异构体或其药学上可接受的盐:
[0043][0044]
在本发明中,r1和r4分别独立地选自氢、卤素、c1-c6烷基、c3-c6环烷基、c2-c6杂烷基、c1-c6烷氧基或c1-c6烷硫基。优选地,r1选自c1-c6烷氧基或c1-c6烷硫基。更优选地,r1为c1-c6烷氧基。根据本发明的一个实施方式,r1为c1-c3烷氧基。优选地,r4选自氢、卤素或c1-c6烷基。更优选地,r4选自氢或c1-c6烷基。根据本发明的一个实施方式,r4为氢。
[0045]
在本发明中,r2选自氢、卤素、c1-c6烷基、c3-c6环烷基、c2-c6杂烷基、c1-c6烷氧基、c1-c6烷硫基或其中,r6选自c1-c6亚烷基、c2-c6杂亚烷基,r5选自氢、卤素、c1-c6烷基、c2-c6杂烷基。优选地,r2为更优选地,r2为其中,r6为c1-c6亚烷基,r5为氢或c1-c6烷基。根据本发明的一个实施方式,r6为c1-c3亚烷基,r5为c1-c3烷基。
[0046]
在本发明中,r3选自氢、c1-c6烷基、c3-c6环烷基、c2-c6杂烷基、c1-c6烷氧基、c1-c6烷硫基或c1-c6酯基。优选地,r3为c1-c6酯基。更优选地,r3为r7选自氢、c1-c6烷基、c3-c6环烷基、c2-c6杂烷基。根据本发明的一个实施方式,r3为r7为c1-c6烷基。
[0047]
在本发明中,卤素的实例包括但不限于氟、氯、溴、碘。
[0048]
在本发明中,c1-c6烷基可以包括但不限于直链烷基或支链烷基;优选为c1-c3烷基,更优选为c1-c3直链烷基。c1-c6烷基的实例包括但不限于甲基、乙基、正丙基、异丙基、正丁基、异丁基、叔丁基、正戊基、异戊基、新戊基、己基等。此外,本发明的c1-c6烷基可以包括取代的烷基或未取代的烷基。取代的烷基中的取代基可以含有杂原子,例如o、s、n或卤素原子。本发明的卤素原子包括但不限于氟、氯、溴、碘。
[0049]
在本发明中,c2-c6杂烷基可以包括但不限于直链杂烷基或支链杂烷基;优选为c2-c5杂烷基,更优选为c2-c3杂烷基。本发明的杂烷基是指烷基链上的碳原子被其他杂原子取代形成的基团。上述杂原子包括o、s或n,优选包括o或s。本发明的c2-c6杂烷基具体的
实例包括但不限于-ch
2-o-ch3、-ch
2-o-ch2ch3、-ch
2-o-ch(ch3)ch3、-ch
2-s-ch3、-ch
2-s-ch2ch3、-ch
2-s-ch(ch3)ch3。
[0050]
在本发明中,c2-c6亚杂烷基可以包括但不限于直链亚杂烷基或支链亚杂烷基;优选为c2-c5亚杂烷基,更优选为c2-c3亚杂烷基。本发明的亚杂烷基是指亚烷基链上的碳原子被其他杂原子取代形成的基团。上述杂原子包括o、s或n,优选包括o或s。本发明的c2-c6亚杂烷基具体的实例包括但不限于-ch
2-o-ch
2-、-ch
2-o-ch2ch
2-、-ch
2-o-ch(ch3)ch
2-、-ch
2-s-ch
2-、-ch
2-s-ch
2-ch
2-、-ch
2-s-ch(ch3)ch
2-。
[0051]
在本发明中,c3-c6环烷基可以包括取代的环烷基和未取代的环烷基;优选为c5-c6环烷基,更优选为c5环烷基。本发明的c3-c6环烷基具体的实例包括但不限于:环丙基、环丁基、环戊基、环己基、3-甲基环戊基、3-甲基环己基、3-乙基环己基,优选为环戊基、环己基。
[0052]
在本发明中,c1-c6烷氧基可以包括但不限于直链烷氧基或支链烷氧基;优选为c1-c3烷氧基,更优选为c1-c3直链烷氧基。c1-c6烷氧基的实例包括但不限于甲氧基、乙氧基、正丙氧基、异丙氧基、正丁氧基、异丁氧基、叔丁氧基、正戊氧基、异戊氧基、新戊氧基、己氧基等。
[0053]
在本发明中,c1-c6烷硫基可以包括但不限于直链烷硫基或支链烷硫基;优选为c1-c3烷硫基,更优选为c1-c3直链烷硫基。c1-c6烷硫基的实例包括但不限于甲硫基、乙硫基、正丙硫基、异丙硫基、正丁硫基、异丁硫基、叔丁硫基、正戊硫基、异戊硫基、新戊硫基、己硫基等。
[0054]
优选地,本发明的醌式喹啉类化合物具有手性结构。4位上的碳为s构型。
[0055]
在本发明中,药学上可接受的盐选自盐酸盐、苯磺酸盐、甲苯磺酸盐、甲磺酸盐、酒石酸盐、苹果酸盐、马来酸盐、富马酸盐、氢溴酸盐、硫酸盐、乙磺酸盐中的一种或多种。
[0056]
本发明的醌式喹啉类化合物可以选自如下化合物之一:
[0057][0058]
优选地,本发明的醌式喹啉类化合物选自如下化合物之一:
[0059][0060]
《制备方法》
[0061]
本发明的醌式喹啉类化合物的制备方法包括如下步骤:(1)山沉香醇提浸膏分离
的步骤;(2)第一流分分离的步骤;(3)第二流分分离的步骤;(4)第三流分分离的步骤;(5)第四流分分离的步骤;和任选地,(6)化学修饰的步骤。在某些实施方式中,还可以包括制备山沉香醇提浸膏的步骤。
[0062]
制备山沉香醇提浸膏的步骤
[0063]
将山沉香采用乙醇水溶液提取,将所得提取液浓缩,得到山沉香醇提浸膏。具体地,将山沉香依次采用第一乙醇水溶液和第二乙醇水溶液提取,分别得到第一提取液和第二提取液。将第一提取液和第二提取液合并,然后浓缩,得到山沉香醇提浸膏。山沉香可以为粉碎后的山沉香。
[0064]
第一乙醇水溶液的浓度为90~97vol%;优选为95vol%。以35.0kg山沉香为基准,第一乙醇水溶液的用量为10~40l;优选为20~30l。
[0065]
第二乙醇水溶液的浓度为75~85vol%;优选为80vol%。以35.0kg山沉香为基准,第二乙醇水溶液的用量为10~40l;优选为20~30l。
[0066]
每次提取时间为0.5~3h;优选为1~2h;更优选为1.5h。
[0067]
提取方式可以为回流提取。浓缩方式可以为减压浓缩。
[0068]
山沉香醇提浸膏分离的步骤
[0069]
将山沉香醇提浸膏采用正相硅胶色谱柱洗脱,结合正相薄层色谱检识,得到流分a-z。流分j或k为第一流分。
[0070]
洗脱溶剂依次采用石油醚-乙酸乙酯-甲醇系统、二氯甲烷-乙酸乙酯-甲醇系统和二氯甲烷-甲醇系统。洗脱方式为梯度洗脱。
[0071]
石油醚-乙酸乙酯-甲醇系统的洗脱条件为:依次采用体积比为(8~12):1:0、(3~7):1:0、(0.8~1.5):1:0、(0.8~1.5):1:(0.1~0.3)、(0.8~1.5):1:(0.8~1.5)和0:0:1的石油醚、乙酸乙酯和甲醇的混合物进行洗脱。优选地,依次采用体积比为10:1:0、5:1:0、1:1:0、1:1:0.2、1:1:1和0:0:1的石油醚、乙酸乙酯和甲醇的混合物进行洗脱。
[0072]
二氯甲烷-乙酸乙酯-甲醇系统的洗脱条件为:依次采用体积比为(18~22):1:0、(8~12):1:0、(0.8~1.5):1:0、(0.8~1.5):1:(0.1~0.3)、(0.8~1.5):1:(0.8~1.5)、0:0:1的二氯甲烷、乙酸乙酯和甲醇的混合物进行洗脱。优选地,依次采用体积比为20:1:0、10:1:0、1:1:0、1:1:0.2、1:1:1和0:0:1的二氯甲烷、乙酸乙酯和甲醇的混合物进行洗脱。
[0073]
二氯甲烷-甲醇系统的洗脱条件为:依次采用体积比为(18~22):1、(8~12):1、(3~7):1、(0.8~1.5):1、0:1的二氯甲烷和甲醇的混合物进行洗脱。优选地,依次采用体积比为20:1、10:1、5:1、1:1和0:1的二氯甲烷和甲醇的混合物进行洗脱。
[0074]
第一流分分离的步骤
[0075]
将第一流分采用葡聚糖凝胶色谱柱洗脱,得到第二流分。根据本发明的一个实施方式色谱柱为葡聚糖凝胶lh-20柱。
[0076]
洗脱溶剂为二氯甲烷和甲醇。洗脱方式为等度洗脱。二氯甲烷和甲醇的体积比为1:(2~6);优选为1:4。
[0077]
当第一流分为流分j时,得到流分ja-jd。以jc作为第二流分。
[0078]
当第一流分为流分k时,得到流分ka-kc。以kc作为第二流分。
[0079]
第二流分分离的步骤
[0080]
将第二流分采用ods色谱柱洗脱,得到第三流分。
[0081]
洗脱溶剂为甲醇和水。洗脱方式为梯度洗脱。
[0082]
洗脱条件为:依次采用体积比为(55~70):(30~45)、100:0的甲醇和水的混合物进行洗脱。在某些实施方式中,依次采用体积比为65:35、100:0的甲醇和水的混合物进行洗脱。在另一些实施方式中,依次采用体积比为60:40、100:0的甲醇和水的混合物进行洗脱。
[0083]
当第二流分为流分jc时,得到jc1-jc5。以jc4作为第三流分。
[0084]
当第二流分为流分kc时,得到kc1-kc6。以kc4作为第三流分。
[0085]
第三流分分离的步骤
[0086]
将第三流分采用葡聚糖凝胶色谱柱洗脱,得到第四流分。根据本发明的一个实施方式色谱柱为葡聚糖凝胶lh-20柱。
[0087]
溶剂为甲醇。洗脱方式为等度洗脱。
[0088]
当第三流分为流分jc4时,得到jc4a-jc4c。以jc4b作为第四流分。
[0089]
当第三流分为流分kc4时,得到kc4a-kc4d。以kc4c作为第四流分。
[0090]
第四流分分离的步骤
[0091]
将第四流分采用半制备液相色谱仪分离,得到醌式喹啉类化合物。
[0092]
流动相为甲醇水溶液。甲醇水溶液的浓度为50~70vol%;优选为55~65vol%。流速为1~8ml/min。在某些实施方式中,流速为2~4ml/min。在另一些实施方式中,流速为5~7ml/min。
[0093]
在某些实施方式中,将流分jc4b采用半制备液相色谱仪,以60~70vol%甲醇水溶液作为流动相分离,流速为5.0~7.0ml/min,得到jc4b1-jc4b6。将流分jc4b3采用半制备液相色谱仪,以50~58vol%甲醇水溶液作为流动相分离,流速为2.0~4.0ml/min,得到化合物d。优选地,将流分jc4b采用半制备液相色谱仪,以65vol%甲醇水溶液作为流动相分离,流速为6.0ml/min,得到jc4b1-jc4b6。将流分jc4b3采用半制备液相色谱仪,以55vol%甲醇水溶液作为流动相分离,流速为3.0ml/min,得到化合物d。
[0094]
在另一些实施方式中,将流分kc4c采用半制备液相色谱仪,以55~60vol%甲醇水溶液作为流动相分离,流速为2.0~4.0ml/min,得到化合物c。优选地,将流分kc4c采用半制备液相色谱仪,以58vol%甲醇水溶液作为流动相分离,流速为3.0ml/min,得到化合物c。
[0095]
化学修饰的步骤
[0096]
将分离得到的化合物采用本领域常规的方法对取代基进行化学修饰,得到醌式喹啉类化合物。
[0097]
《用途》
[0098]
本发明的醌式喹啉类化合物具有抗肿瘤和抗菌活性,因而本发明提供上述醌式喹啉类化合物在制备抗肿瘤和/或抗菌药物中的用途。在某些实施方式中,本发明提供了上述醌式喹啉类化合物在制备抗肿瘤药物中的用途。在另一些实施方式中,本发明提供了上述醌式喹啉类化合物在制备抗菌药物中的用途。
[0099]
在本发明中,肿瘤的实例包括但不限于肝癌、乳腺癌、前列腺癌、皮肤癌、头颈癌、肺癌、食道癌、宫颈癌、胰腺癌、结肠癌、肾癌、输尿管癌、膀胱癌。优选地,肿瘤为乳腺癌、肝癌、前列腺癌中的一种或多种。更优选地,肿瘤为乳腺癌、肝癌中的一种或多种。
[0100]
在本发明中,菌选自枯草芽孢杆菌、白色念珠菌、金黄色葡萄球菌、铜绿假单胞菌、沙门菌中的一种或多种。优选地,菌为枯草芽孢杆菌。
[0101]
实施例1
[0102]
将35.0kg山沉香粉碎,然后依次采用25l 95vol%的乙醇水溶液和25l 80vol%的乙醇水溶液回流提取,每次提取时间为1.5h,分别得到第一提取液和第二提取液。合并第一提取液和第二提取液,然后减压浓缩,得到山沉香醇提浸膏。
[0103]
将山沉香醇提浸膏采用正相硅胶色谱柱,依次以石油醚-乙酸乙酯-甲醇系统(10:1:0
→
5:1:0
→
1:1:0
→
1:1:0.2
→
1:1:1
→
0:0:1),二氯甲烷-乙酸乙酯-甲醇系统(20:1:0
→
10:1:0
→
1:1:0
→
1:1:0.2
→
1:1:1
→
0:0:1),二氯甲烷-甲醇系统(20:1
→
10:1
→
5:1
→
1:1
→
0:1)作为溶剂梯度洗脱,结合正相薄层色谱检识,得到流分a-z。
[0104]
实施例2
[0105]
将实施例1得到的流分j采用葡聚糖凝胶lh-20柱,以体积比为1:4的二氯甲烷和甲醇作为溶剂洗脱,得到4个流分ja-jd。
[0106]
将jc流分采用ods色谱柱,以甲醇-水系统(65:35
→
100:0)作为溶剂梯度洗脱,得到5个流分jc1-jc5。
[0107]
将jc4流分采用葡聚糖凝胶lh-20柱,以甲醇作为溶剂洗脱,得3个流分jc4a-jc4c。
[0108]
将jc4b流分采用半制备液相色谱仪,以65vol%甲醇水溶液作为流动相分离,流速为6.0ml/min,得到6个流分jc4b1-jc4b6。
[0109]
将jc4b3流分采用半制备液相色谱仪,以55vol%甲醇水溶液作为流动相分离,流速为3.0ml/min,得到化合物d(10.0mg,tr=28.2min)。
[0110]
实施例3
[0111]
将实施例1得到的流分k采用葡聚糖凝胶lh-20柱,以体积比为1:4的二氯甲烷和甲醇作为溶剂洗脱,得到3个流分ka-kc。
[0112]
将流分kc采用ods色谱柱,以甲醇-水系统(60:40
→
100:0)作为溶剂梯度洗脱,得到6个流分kc1-kc6。
[0113]
将流分kc4采用葡聚糖凝胶lh-20柱,以甲醇作为溶剂洗脱,得4个流分kc4a-kc4d。
[0114]
将流分kc4c采用半制备液相色谱仪,以58vol%甲醇水溶液作为流动相分离,流速为3.0ml/min,得到化合物c(1.5mg,tr=31.5min)。
[0115]
化合物c和化合物d的1h-nmr(500mhz,cdcl3)和
13
c-nmr(125mhz,cdcl3)数据如下表所示:
[0116][0117]
化合物c:经hr-esi-ms检测,其准分子离子峰[m
–
h]
–
372.1063(计算值372.1078),结合
13
c-nmr数据确定其分子式为c
19h17
no7,不饱和度为12。1h-nmr数据显示分子中有四个烯氢,一个次甲基,一个亚甲基,三个甲氧基,一个伯胺氢。
13
c-nmr数据显示其分子中含有19个碳原子,其中包括4个羰基碳,10个不饱和碳,一个次甲基,一个亚甲基,三个甲氧基。结合hsqc与hmbc图谱对其1h和
13
c nmr数据进行了归属,如表1所示。以上信息推测化合物c为生物碱类,且结构中含有一个醌式片段。通过计算ecd的方法确定其绝对构型为4s。化合物c的结构如下所示:
[0118][0119]
化合物d:经hr-esi-ms检测,其准分子离子峰[m
–
h]
–
386.1222(计算值386.1234),结合
13
c-nmr数据确定其分子式为c
20h19
no7,不饱和度为12。1h-nmr和
13
c-nmr数据与化合物c的数据对比发现,化合物d中c-13位由乙氧基取代,由此确定了化合物d的平面结构,结合hsqc与hmbc图谱对其1h和
13
c nmr数据进行了归属,如表1所示。通过与化合物c的ecd图谱进行比对,该化合物的绝对构型得以确定为4s。化合物d的结构如下所示:
[0120][0121]
实验例1-mtt法测试化合物对癌症细胞增殖的影响
[0122]
取对数生长期培养的3种癌细胞mcf-7、hepg2和pc-3m-ie8,分别用含10%胎牛血清的新鲜1640培养基培养,将细胞密度调至5
×
104个/ml分别接种于96孔板中。将孔板放在含5%co2,37℃下培养24h,然后分别加入0μm、1μm、5μm、10μm、15μm、20μm和30μm的化合物c或化合物d。药物预处理48h后吸上培养基,然后加上100μl 500μg/ml的mtt溶液于培养箱中继续培养4h。小心吸弃mtt,每孔加入150μl的dmso,然后将孔板在平板振荡器上振荡10分钟。将96孔板置于酶标仪中,检测570nm处的od值。利用酶标仪及相应软件进行数据处理,按如下公式计算细胞存活率和抑制率:细胞存活率%=样品组od平均值/空白对照组od平均值
×
100%。抑制率%=100%-细胞存活率%。所得结果如图2-6所示。
[0123]
化合物c对hepg2和mcf-7细胞具有明显的抑制活性。当化合物c的浓度为30μm时,对mcf-7细胞的抑制率为94%,对hepg2细胞的抑制率为93%。化合物c对mcf-7细胞的ic
50
为5.0μmol/l,对hepg2细胞的ic
50
为9.1μmol/l。
[0124]
化合物d对mcf-7、hepg2和pc-3m-ie8细胞具有明显的抑制活性。当化合物d的浓度为30μm时,对mcf-7细胞的抑制率为93%,对hepg2细胞的抑制率为91%,对pc-3m-ie8细胞的抑制率为71%。化合物d对mcf-7细胞的ic
50
为10.3μmol/l,对hepg2细胞的ic
50
为12.1μmol/l,对pc-3m-ie8细胞的ic
50
为18.1μmol/l。
[0125]
实验例2-测试化合物对枯草芽孢杆菌和白色念珠菌的影响
[0126]
分别将枯草芽孢杆菌atcc6633(购自中国普通微生物菌种保藏管理中心)和白色
念珠菌atcc10231(购自中国普通微生物菌种保藏管理中心)在酵母提取物-蛋白胨-葡萄糖培养基(ypd)中生长至对数中期,然后用相同的培养基稀释至5
×
105cfu/ml的密度,得到枯草芽孢杆菌菌液和白色念珠菌菌液。将枯草芽孢杆菌菌液和白色念珠菌菌液分别形成一系列含有梯度浓度的化合物c的枯草芽孢杆菌菌液和白色念珠菌菌液(化合物c的含量为1~87μg/ml)和一系列含有梯度浓度的化合物d的枯草芽孢杆菌菌液和白色念珠菌菌液(化合物d的含量为1~87μg/ml)。将含有化合物c的枯草芽孢杆菌菌液、含有化合物c的白色念珠菌菌液、含有化合物d的枯草芽孢杆菌菌液、含有化合物d的白色念珠菌菌液分别以0.2ml/孔分配到无菌96孔板中。在37℃下孵育17小时后,通过测量和比较空白对照和测试孔的光学多样性来确定最小抑制浓度(mic),并记录。抗菌阳性对照为利福平。所有试验均一式三份进行。
[0127]
结果显示化合物c和化合物d对枯草芽孢杆菌具有影响,对白色念珠菌影响不明显。化合物c和化合物d对枯草芽孢杆菌具有明显的抗菌活性,且最小抑菌浓度(mic)分别为10.88μg/ml和5.31μg/ml。阳性对照药利福平最小抑菌浓度为0.04μg/ml。
[0128]
本发明并不限于上述实施方式,在不背离本发明的实质内容的情况下,本领域技术人员可以想到的任何变形、改进、替换均落入本发明的范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1