用于生产反式-4-羟基脯氨酸的基因工程菌及其构建方法与应用
1.本发明涉及化合物生物技术及发酵工程技术生产领域,尤其是一种用于生产反式-4-羟基脯氨酸的基因工程菌及其构建方法与应用。
背景技术:
2.反式-4-羟脯氨酸(trans-4-hydroxy-l-proline),羟脯氨酸由脯氨酸羟基化得到,关键酶为脯氨酸羟化酶,它是动物结构蛋白(如胶原蛋白和弹性蛋白)的天然成分,被广泛应用于医药、食品、饲料及化妆品领域。现如今,随着人们对羟脯氨酸功能研究的逐渐深入,其市场需求还在不断扩大。
3.反式-4-羟脯氨酸的生产方法主要有蛋白水解法、化学合成法和微生物发酵法,但前两种方法使用大量的酸碱试剂及有机试剂,存在提取困难、污染严重、价格昂贵等问题,无法满足生产的需求。
4.微生物发酵法是以廉价的葡萄糖为碳源进行微生物生产的高效方法,具有绿色环保、操作简单等优点,但往往发酵过程难以控制。
技术实现要素:
5.本发明所要解决的技术问题在于提供一种大肠杆菌菌株。
6.本发明所要解决的另一技术问题在于提供上述大肠杆菌菌株的构建方法。
7.本发明所要解决的另一技术问题在于提供上述大肠杆菌菌株的应用。
8.为解决上述技术问题,本发明的技术方案是:
9.一种用于生产反式-4-羟基脯氨酸的基因工程菌,命名为e.coli hyp,所述基因工程菌e.coli hyp是在野生型大肠杆菌的染色体基因组上敲除脯氨酸脱氢酶基因;染色体基因组上敲除脯氨酸转运蛋白基因;染色体基因组上敲除脯氨酸转运体基因;染色体基因组上敲除dna结合转录抑制因子基因;染色体基因组上敲除异柠檬酸裂解酶基因;并分别在基因组mbha位点和yghx位点上整合密码子优化(按照大肠杆菌密码子偏好性进行优化)后的谷氨酸激酶基因prob(ncbi-gene id:946425),并由ptrc启动子(具有序列表seq id no.9所示核苷酸序列)启动;在基因组yciq位点上整合吡咯啉-5-羧酸还原酶基因proc(ncbi-gene id:945034);并由同一个ptrc启动子启动;分别在基因组ycit位点和ycgh位点上整合密码子优化后的谷氨酸半醛脱氢酶基因proa(ncbi-gene id:946680),并由ptrc启动子启动;在基因组ygay位点上整合密码子优化后的脯氨酸羟化酶基因hyp,并由ptrc启动子启动;在基因组yeep位点上整合密码子优化后的解酮酶基因xfp(ncbi-gene id:56674845))并由同一个ptrc启动子启动;在基因组yjip位点上分段整合吡啶核苷酸转氢酶基因pnta(ncbi-geneid:946628)和pntb(ncbi-gene id:886830),并由同一个ptrc启动子启动;在基因组yjiv位点上整合透明颤菌血红蛋白基因vhb(ncbi-gene id:cp020373.1),并由同一个ptrc启动子启动;在基因组yjgx位点上整合脯氨酸转运蛋白基因cgl2622(ncbi-gene id:
cp025533.1),并由同一个ptrc启动子启动。
10.优选的,上述用于生产反式-4-羟基脯氨酸的基因工程菌,所述野生型大肠杆菌为大肠杆菌e.coli w3110(atcc 27325)。
11.优选的,上述用于生产反式-4-羟基脯氨酸的基因工程菌,所述脯氨酸羟化酶基因为密码子优化后基因hyp。
12.上述用于生产反式-4-羟基脯氨酸的基因工程菌的构建方法,采用crispr/cas9介导的基因编辑技术对e.coli w3110染色体基因组进行定向改造,具体步骤如下:
13.(1)为了进一步增加脯氨酸的供应,阻断脯氨酸反馈抑制途径,对脯氨酸脱氢酶编码基因puta进行敲除;
14.(2)为了减弱脯氨酸向胞内积累,阻断脯氨酸向胞内运输,对脯氨酸转运蛋白编码基因prop进行敲除;
15.(3)为了减弱脯氨酸的内排途径,阻断脯氨酸向胞内运输,对脯氨酸转运体编码基因putp进行敲除;
16.(4)为了减少对ptrc启动子的抑制,增强其表达效果,对dna结合转录抑制因子编码基因laci进行敲除;
17.(5)为了减弱α-酮戊二酸分支代谢途径,阻断乙醛酸代谢,对异柠檬酸裂解酶编码基因acea进行敲除;
18.(6)为了进一步加强脯氨酸合成途径,将密码子优化后的谷氨酸激酶编码基因prob整合至大肠杆菌基因组的mbha和yghx位点,并用ptrc启动子控制转录;
19.(7)为了进一步加强脯氨酸合成代谢通路,将吡咯啉-5-羧酸还原酶编码基因proc整合至大肠杆菌基因组的yciq位点,用同一个ptrc启动子控制转录;
20.(8)为了进一步加强脯氨酸合成途径,将密码子优化后的谷氨酸半醛脱氢酶编码基因proa整合至大肠杆菌基因组的ycit和ycgh位点,并用ptrc启动子控制转录;
21.(9)为了加强反式-4-羟基脯氨酸合成途径,将密码子优化后的脯氨酸羟化酶编码基因hyp整合至大肠杆菌基因组的yghx位点,用同一个ptrc启动子控制转录;
22.(10)为了加强谷氨酸合成途径,将密码子优化后的解酮酶编码基因xfp整合至大肠杆菌基因组的yeep位点,用同一个ptrc启动子控制转录;
23.(11)为了加强细胞内nadph供应,将吡啶核苷酸转氢酶编码基因pnta和pntb整合至大肠杆菌基因组的yjip位点,用同一个ptrc启动子控制转录;
24.(12)为了加强细胞内氧的代谢途径,将血红颤菌蛋白编码基因vhb整合至大肠杆菌的yjiv位点,用同一个ptrc启动子控制转录;
25.(13)为了加强脯氨酸向细胞外转运,将脯氨酸转运蛋白编码基因cgl2622整合至大肠杆菌的yjgx位点,用同一个ptrc启动子控制转录。
26.上述基因工程菌在生产反式-4-羟基脯氨酸方面的应用。
27.优选的,上述基因工程菌的应用,利用所述基因工程菌进行摇瓶发酵:按总体积10%-15%的接种量接种菌种活化后制备的种子液到装有发酵培养基的三角瓶中(终体积为30ml),九层纱布封口,37℃,220r/min振荡培养,发酵过程中通过补加25%的氨水(v/v)维持ph在7.0-7.2;添加60%(m/v)葡萄糖溶液维持发酵进行(以苯酚红做指示剂,发酵液颜色不再变化时即视为缺糖,缺糖时补加1-2ml 60%(m/v)葡萄糖溶液),发酵周期30-32h。
28.优选的,上述基因工程菌的应用,所述发酵培养基的组成为:葡萄糖20g/l,酵母粉6g/l,蛋白胨1g/l,柠檬酸1.5g/l,(nh4)2so
4 1g/l,mgso4
·
7h2o 1.5g/l,kh2po
4 4.0g/l,mnso
4 10mg/l,谷氨酸1g/l,蛋氨酸0.2mg/l,α-酮戊二酸0.5g/l,上述成分称量固体后溶解于1l水中,在4℃保存。
29.上述基因工程菌e.coli hyp利用摇瓶发酵30-32h后反式-4-羟基脯氨酸的产量可达22-25g/l。
30.优选的,上述基因工程菌的应用,利用所述基因工程菌进行发酵罐发酵:
31.(1)种子培养:取适量无菌水重悬活化斜面中的一代种子,将菌悬液接入种子培养基中,ph维持在7.0-7.2,温度维持在36℃,溶氧在30%-60%之间,培养5.8-6h;
32.(2)发酵培养:等待种子菌体量od600至20-25,按照20%接种量接入发酵培养基,开始发酵,发酵过程中控制ph稳定在7.0-7.2,温度维持在36℃,溶氧在30%-60%之间;当培养基中的葡萄糖消耗完之后,流加80%(m/v)的葡萄糖溶液,维持发酵培养基中的葡萄糖浓度在0.1-1g/l(残糖仪测量);发酵周期45h。
33.优选的,上述基因工程菌的应用,所述发酵培养基的组成为:葡萄糖20g/l,酵母粉6g/l,蛋白胨1g/l,柠檬酸1.5g/l,(nh4)2so
4 1g/l,mgso4
·
7h2o 1.5g/l,kh2po
4 4.0g/l,mnso
4 10mg/l,谷氨酸1g/l,蛋氨酸0.2mg/l,α-酮戊二酸0.5g/l,上述成分称量固体后溶解于1l水中,在4℃保存。
34.上述基因工程菌e.coli hyp利用5l发酵罐发酵45h后反-4-羟基脯氨酸的产量可达129.2g/l,糖酸转化率为36.3%。
35.有益效果:
36.本发明所述用于生产反式-4-羟基脯氨酸的基因工程菌,弃除了以往菌种改造中携带质粒、发酵过程难以控制的缺点,以大肠杆菌作为底盘细胞,构建了一株不含质粒、从头合成高产反式-4-羟基脯氨酸的基因工程菌e.coli hyp,与现有反式-4-羟基脯氨酸生产菌相比,该基因工程菌具有遗传稳定性好、发酵产率高、糖酸转化率高等优势,所述基因工程菌以葡萄糖等廉价碳源为底物从头高效合反式-4-羟基脯氨酸,反式-4-羟基脯氨酸产量可高达129.2g/l,糖酸转化率为36.3%,具有很好的工业应用前景;该基因工程菌构建方法简单,首先对大肠杆菌中谷氨酸代谢网络中与反式-4-羟基脯氨酸的合成途径进行分析重构,加强了反式-4-羟基脯氨酸合成和积累;之后引入密码子优化后的指孢囊菌脯氨酸羟化酶基因,进一步加强反式-4-羟基脯氨酸合成,适合规模化工业生产的需要。
附图说明
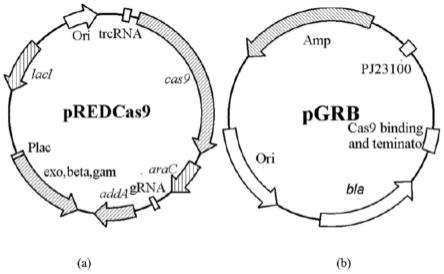
37.图1:(a)pred/cas9质粒图谱,(b)pgrb质粒图谱。
38.图2:一种产生反式-4-羟基脯氨酸工程菌的构建方法图解。
39.图3:puta基因敲除片段构建及验证电泳图。其中:m:1kb dna marker;1:上游同源臂;2:下游同源臂;3:重叠片段;4:阳性菌鉴定片段;5:原菌对照。
40.图4:prop基因敲除片段构建及验证电泳图。其中:m:1kb dna marker;1:上游同源臂;2:下游同源臂;3:重叠片段;4:阳性菌鉴定片段;5:原菌对照。
41.图5:putp基因敲除片段构建及验证电泳图。其中:m:1kb dna marker;1:上游同源臂;2:下游同源臂;3:重叠片段;4:阳性菌鉴定片段;5:原菌对照。
editing.metabolic engineering,2015,31:13-21.),该方法涉及的工程质粒pred/cas9、pgrb图谱见图1。其中pred/cas9携带grna表达质粒pgrb的消除系统、λ噬菌体的red重组系统、cas9蛋白表达系统以及奇霉素抗性(工作浓度:100mg/l),32℃培养;pgrb以puc18为骨架,包括启动子j23100、grna-cas9结合区域序列和终止子序列以及氨苄青霉素抗性(工作浓度:50mg/l),37℃培养,所述质粒pgrb具有序列表seq id no.10所示核苷酸序列,所述质粒cas9具有序列表seq id no.11所示核苷酸序列。
62.该方法的具体步骤如下:
63.1.1pgrb质粒构建
64.采用包含靶序列的dna片段与线性化的pgrb载体片段重组的方法构建得到pgrb质粒,目的是为了转录相应的grna,与cas9蛋白形成的复合体,并通过碱基配对和pam识别目的基因靶位点,实现目的dna双链断裂。
65.1.1.1靶序列设计
66.使用crispr rgen tools设计靶序列(pam:5'-ngg-3')
67.1.1.2包含靶序列的dna片段的制备
68.设计引物:5'-线性化载体末端序列(15bp)-靶序列-线性化载体末端序列(15bp)-3',及其反向互补的引物,利用pcr退火程序得到dna双链片段。反应条件:预变性95℃,5min;退火30-50℃,1min。退火体系如下表1:
69.表1退火体系
[0070][0071]
1.1.3线性载体的制备
[0072]
载体的线性化采用反向pcr扩增的方法。
[0073]
1.1.4重组连接靶序列与线性化载体
[0074]
使用ii one step cloning kit重组酶重组连接靶序列与线性化的pgrb载体(反应体系见表2),插入片段见1.1.2,线性化克隆载体见1.1.3,将得到了质粒化转至e.coli dh5α感受态细胞后筛选阳性转化子,菌株纯化后摇管扩大培养,使用试剂盒提取质粒,得到含靶序列的pgrb质粒。质粒重组体系如下表2:
[0075]
表2质粒重组体系
[0076][0077]
1.2重组dna片段的制备
[0078]
重组dna片段以整合位点的上下游同源臂及待整合的基因片段组成(ptrc启动子整合于重叠片段中,仅作敲除目的则不需要目的片段,所述ptrc启动子具有序列表seq id no.9所示核苷酸序列)。以待敲除基因/目的基因的上同源臂的上游引物与下同源臂的下引
物为扩增/重叠引物,以大肠杆菌基因组为模板,利用pcr扩增体系(如表3)得到同源臂/目的基因的dna片段。以待敲除基因的上同源臂的上游引物与下同源臂的下游引物为重叠引物,以待整合基因为模板,利用pcr重叠体系(表4)制备重组片段。
[0079]
表3 hs酶pcr扩增体系
[0080][0081]
表4重叠pcr扩增体系
[0082][0083]
pcr反应条件(宝生物primestar hs酶):预变性(95℃)5min;变性(98℃)10s,退火((tm-3/5)℃)15s,72℃延伸(酶活力1min延伸约1kb)进行30轮循环;72℃继续延伸10min;维持(4℃)。
[0084]
1.3质粒和重组dna片段的转化
[0085]
1.3.1pred/cas9的转化
[0086]
利用电转的方法将pred/cas9质粒电转至目的菌株的电转感受态中,复苏培养后涂布在含奇霉素抗性的lb固体平板上,32℃培养12h。抗性平板上挑选单菌落用鉴定引物进行菌落pcr验证,筛选阳性转化子。菌落pcr体系如下表5:
[0087]
表5菌落pcr体系
[0088][0089]
1.3.2含pred/cas9的目的菌株电转化感受态制备
[0090]
32℃培养至od600nm=0.1~0.2时,添加0.1m的iptg(使其终浓度为0.1mm),目的为诱导pred/cas9质粒上的重组酶表达。其他操作无特殊要求。
[0091]
1.3.3pgrb和重组dna片段的转化
[0092]
将pgrb和供体dna片段同时电转化至含有pred/cas9的电转感受态细胞中。将电转
化后复苏培养的菌体涂布于含氨苄青霉素和奇霉素的lb平板上,32℃过夜培养。用上游同源臂上游引物和下游同源臂的下游引物,或设计专门的鉴定引物,进行菌落pcr验证,筛选阳性重组子并保菌。
[0093]
1.4质粒的消除
[0094]
1.4.1pgrb质粒的消除
[0095]
将阳性重组子置于含有0.2%阿拉伯糖的lb培养基中培养12h,取适量菌液涂布于含有奇霉素抗性的lb平板上,32℃培养12h。使用含有氨苄青霉素和奇霉素抗性的lb平板(单抗平板,对照筛选阳性菌株),挑选氨苄青霉素平板不生长,奇霉素抗性平板生长的单菌落保菌。
[0096]
1.4.2pred/cas9质粒的消除
[0097]
将阳性重组子转接到无抗性的lb液体培养基中,42℃培养12h,取适量菌液涂布于无抗性的lb平板上,37℃培养12h。使用含有奇霉素抗性和无抗性的lb平板(单抗平板,对照筛选阳性菌株),挑选奇霉素抗性平板不生长,无抗性平板生长的单菌落保菌。
[0098]
2.菌株构建过程中所涉及的引物如下表6:
[0099]
表6引物序列
[0100]
[0101]
[0102]
[0103]
[0104][0105]
上述菌株构建的具体过程如下:
[0106]
3.1基因puta的敲除
[0107]
以提取并稀释至可用浓度的大肠杆菌w3110基因组为模板,用引物puta-q-1和pu-taq-2,puta-q-3和puta-q-4分别进行pcr扩增,得到上游同源臂和下游同源臂,大小分别为446bp和465bp,以回收的上下游同源臂为模板,用引物puta-q-1和puta-q-4进行重叠pcr,得到敲除基因puta所需的回补片段δputa,大小在1030bp。之后,将引物pgrb-puta-s和pgrb-puta-a退火制得的dna片段与质粒pgrb线载连接,构建pgrb-puta。制备e.coli w3110/pred-cas9的感受态细胞,按照1.3和1.4所示的方法操作,最终获得菌株e.coli hyp-1。过程中片段扩增结果和阳性菌株的pcr验证的电泳图见图3,各片段大小均与理论值大小一致。
[0108]
3.2基因prop的敲除
[0109]
以提取并稀释至可用浓度的大肠杆菌w3110基因组为模板,用引物prop-q-1和prop-q-2,prop-q-3和prop-q-4分别进行pcr扩增,得到上游同源臂和下游同源臂,大小分
别为464bp和343bp,以回收的上下游同源臂为模板,用引物prop-q-1和prop-q-4进行重叠pcr,得到敲除基因prop所需的回补片段δprop,大小在927bp。之后,将引物pgrb-prop-s和pgrb-prop-a退火制得的dna片段与质粒pgrb线载连接,构建pgrb-prop。制备e.coli hyp-1/pred-cas9的感受态细胞,按照1.3和1.4所示的方法操作,最终获得菌株e.coli hyp-2。过程中片段扩增结果和阳性菌株的pcr验证的电泳图见图4,各片段大小均与理论值大小一致。
[0110]
3.3基因putp敲除
[0111]
以提取并稀释至可用浓度的大肠杆菌w3110基因组为模板,用引物putp-q-1和putp-q-2,putp-q-3和putp-q-4分别进行pcr扩增,得到上游同源臂和下游同源臂,大小分别为389bp和439bp,以回收的上下游同源臂为模板,用引物prop-q-1和putp-q-4进行重叠pcr,得到敲除基因putp所需的回补片段δprop,大小在948bp。之后,将引物pgrb-putp-s和pgrb-putp-a退火制得的dna片段与质粒pgrb线载连接,构建pgrb-putp。制备e.coli hyp-2/pred-cas9的感受态细胞,按照1.3和1.4所示的方法操作,最终获得菌株e.coli hyp-3。过程中片段扩增结果和阳性菌株的pcr验证的电泳图见图5,各片段大小均与理论值大小一致。
[0112]
3.4基因laci的敲除
[0113]
以提取并稀释至可用浓度的大肠杆菌w3110基因组为模板,用引物laci-q-1和laci-q-2,laci-q-3和laci-q-4分别进行pcr扩增,得到上游同源臂和下游同源臂,大小分别为454bp和448bp,以回收的上下游同源臂为模板,用引物laci-q-1和laci-q-4进行重叠pcr,得到敲除基因laci所需的回补片段δlaci,大小在1022bp。之后,将引物pgrb-laci-s和pgrb-laci-a退火制得的dna片段与质粒pgrb线载连接,构建pgrb-p。制备e.coli hyp-3/pred-cas9的感受态细胞,按照1.3和1.4所示的方法操作,最终获得菌株e.coli hyp-4。过程中片段扩增结果和阳性菌株的pcr验证的电泳图见图6,各大小均与理论值大小一致。
[0114]
3.5基因acea的敲除
[0115]
以提取并稀释至可用浓度的大肠杆菌w3110基因组为模板,用引物acea-q-1和acea-q-2,acea-q-3和acea-q-4分别进行pcr扩增,得到上游同源臂和下游同源臂,大小分别为558bp和437bp,以回收的上下游同源臂为模板,用引物laci-q-1和laci-q-4进行重叠pcr,得到敲除基因laci所需的回补片段δlaci,大小在1155bp。之后,将引物pgrb-laci-s和pgrb-laci-a退火制得的dna片段与质粒pgrb线载连接,构建pgrb-p。制备e.coli hyp-4/pred-cas9的感受态细胞,按照1.3和1.4所示的方法操作,最终获得菌株e.coli hyp-5。过程中片段扩增结果和阳性菌株的pcr验证的电泳图见图7,各大小均与理论值大小一致。
[0116]
3.6基因prob的整合
[0117]
以大肠杆菌w3110为模板,用引物mbha-up-s和mbha-up-a,mbha-dw-s和mbha-dw-a,prob-trc-s和prob-trc-a进行pcr扩增,得到上下游同源臂和中间目的片段,其理论大小分别在626bp、619bp、1100bp,以回收的上下游同源臂和中间目的片段为模板,用引物mbha-up-s和mbha-dw-a经重叠pcr得到整合需要的目的片段ptrc-prob1理论大小在2468bp。之后,将引物pgrb-mbha-s和pgrb-mbha-a退火制得的dna片段与质粒pgrb线载连接,构建pgrb-ycgh质粒。制备e.coli hyp-5/pred-cas9的感受态细胞,按照1.3和1.4所示的方法操作,最终获得菌株e.coli hyp-6。过程中片段扩增结果和阳性菌株的pcr验证的电泳图见图
a,hyp-t7-s和hyp-t7-a进行pcr扩增,得到上下游同源臂和中间目的片段,其理论大小分别在616bp、617bp、814bp,以回收的上下游同源臂和中间目的片段为模板,用引物yciq-up-s和yciq-dw-a经重叠pcr得到整合需要的目的片段pt7-hyp理论大小在2167bp。之后,将引物pgrb-ygay-s和pgrb-ygay-a退火制得的dna片段与质粒pgrb线载连接,构建pgrb-ygay质粒。制备e.coli hyp-10/pred-cas9的感受态细胞,按照1.3和1.4所示的方法操作,最终获得菌株e.coli hyp-11。过程中片段扩增结果和阳性菌株的pcr验证的电泳图见图11,各片段大小均与理论值大小一致。
[0128]
3.12基因xfp的整合
[0129]
以大肠杆菌w3110为模板,用引物yeep-up-s和yeep-up-a,yeep-dw-s和yeep-dw-a,xfp-trc-s和xfp-trc-a进行pcr扩增,得到上下游同源臂和中间目的片段,其理论大小分别在512bp、514bp、2490bp,以回收的上下游同源臂和中间目的片段为模板,用引物yeep-up-s和yeep-dw-a经重叠pcr得到整合需要的目的片段ptrc-xfp理论大小在3636bp。之后,将引物pgrb-yeep-s和pgrb-yeep-a退火制得的dna片段与质粒pgrb线载连接,构建pgrb-yeep质粒。制备e.coli hyp-9-11/pred-cas9的感受态细胞,按照1.3和1.4所示的方法操作,最终获得菌株e.coli hyp-12。过程中片段扩增结果和阳性菌株的pcr验证的电泳图见图12,各片段大小均与理论值大小一致。
[0130]
3.13基因pntab的整合
[0131]
以大肠杆菌w3110为模板,用引物yjip-up1-s和yjip-up1-a,yjip-dw1-s和yjip-dw1-a,pntab1-trc-s和pntab1-trc,yjip-up2-s和yjip-up2-a,yjip-dw2-s和yjip-dw2-a,进行pcr扩增,分别得到第一段和第二段的上下游同源臂和中间目的片段,其理论大小分别在499bp、463bp、2100bp、1679bp、463bp,以回收的上下游同源臂和中间目的片段为模板,用引物yjip-up1-s和yjip-dw1-a经重叠pcr得到整合需要的第一段目的片段ptrc-pntab1理论大小在3182bp,用引物yjip-up2-s和yjip-dw2-a经重叠pcr得到整合需要的第二段目的片段ptrc-pntab2理论大小在2262bp。之后,将引物pgrb-yjip-s1和pgrb-yjip-a1退火制得的dna片段与质粒pgrb线载连接,构建pgrb-yjip1质粒,引物pgrb-yjip-s2和pgrb-yjip-a2退火制得的dna片段与质粒pgrb线载连接,构建pgrb-yjip2质粒。制备e.coli hyp-12/pred-cas9的感受态细胞,按照1.3和1.4所示的方法操作,最终获得菌株e.coli hyp-13。过程中片段扩增结果和阳性菌株的pcr验证的电泳图见图13-1和图13-2,各片段大小均与理论值大小一致。
[0132]
3.14基因vhb的整合
[0133]
以大肠杆菌w3110为模板,用引物yjiv-up-s和yjiv-up-a,yjiv-dw-s和yjiv-dw-a,vhb-trc-s和vhb-trc-a进行pcr扩增,得到上下游同源臂和中间目的片段,其理论大小分别在557bp、677bp、426bp,以回收的上下游同源臂和中间目的片段为模板,用引物yjiv-up-s和yjiv-dw-a经重叠pcr得到整合需要的目的片段ptrc-vhb理论大小在1780bp。之后,将引物pgrb-yjiv-s和pgrb-yjiv-a退火制得的dna片段与质粒pgrb线载连接,构建pgrb-yjiv质粒。制备e.coli hyp-13/pred-cas9的感受态细胞,按照1.3和1.4所示的方法操作,最终获得菌株e.coli hyp-14。过程中片段扩增结果和阳性菌株的pcr验证的电泳图见图14,各片段大小均与理论值大小一致。
[0134]
3.15基因cgl2622转运蛋白的整合
[0135]
以大肠杆菌w3110为模板,用引物yjgx-up-s和yjgx-up-a,yjgx-dw-s和yjgx-dw-a,2622-trc-s和2622-trc-a进行pcr扩增,得到上下游同源臂和中间目的片段,其理论大小分别在505bp、422bp、1470bp,以回收的上下游同源臂和中间目的片段为模板,用引物yjgx-up-s和yjgx-up-a经重叠pcr得到整合需要的目的片段ptrc-2622理论大小在2397bp。之后,将引物pgrb-2622-s和pgrb-2622-a退火制得的dna片段与质粒pgrb线载连接,构建pgrb-yjgx质粒。制备e.coli hyp-14/pred-cas9的感受态细胞,按照1.3和1.4所示的方法操作,最终获得菌株e.coli hyp-15。过程中片段扩增结果和阳性菌株的pcr验证的电泳图见图15,各片段大小均与理论值大小一致。
[0136]
4.密码子优化前后核苷酸序列
[0137]
4.1基因prob具有序列表seq id no.1所示核苷酸序列;
[0138]
4.2基因proa具有序列表seq id no.2所示核苷酸序列;
[0139]
4.3 hyp具有序列表seq id no.3所示核苷酸序列;
[0140]
4.4 xfp具有序列表seq id no.4所示核苷酸序列;
[0141]
4.5 prob密码子优化后具有序列表seq id no.5所示核苷酸序列;
[0142]
4.6 proa密码子优化后具有序列表seq id no.6所示核苷酸序列;
[0143]
4.7 hyp密码子优化后具有序列表seq id no.7所示核苷酸序列;
[0144]
4.8 xfp密码子优化后具有序列表seq id no.8所示核苷酸序列。
[0145]
实施例2
[0146]
采用实施例1所述的大肠杆菌工程菌利用摇瓶发酵反式-4-羟基脯氨酸
[0147]
1.培养基
[0148]
1.1斜面培养基
[0149]
葡萄糖2g/l,蛋白胨10g/l,酵母浸出粉5g/l,氯化钠2.5g/l,kh2po4 1.0g/l,mgso4 0.2g/l,琼脂粉2.5%,用水溶解并定容至所需体积,用1mol/l氢氧化钠调ph至7.0-7.2,121℃高压蒸汽锅灭菌20min后分装至试管中。
[0150]
1.2种子培养基
[0151]
葡萄糖30g/l,酵母粉6g/l,蛋白胨4g/l,柠檬酸1g/l,(nh4)2 so4 3g/l,mgso4
·
7h2o 0.4g/l,kh2po4 1.5g/l,mnso4 5mg/l,vb
1 0.5mg/l,微量元素混合液1ml/l,谷氨酸0.5g/l,蛋氨酸0.5mg/l,氨基酸粉2g/l,苯酚红:需定容体积的2%,用naoh调ph至7.0-7.2,消泡剂1滴,其余为水;所述微量元素混合液组分含量为:钼酸铵0.28mg/l,mnso4·
h2o 0.5mg/l,cuso4·
7h2o 0.5mg/l。
[0152]
上述成分称量固体后溶解于1l水中,在4℃保存。
[0153]
1.3发酵培养基
[0154]
葡萄糖20g/l,酵母粉6g/l,蛋白胨1g/l,柠檬酸1.5g/l,(nh4)2so4 1g/l,mgso4·
7h2o 1.5g/l,kh2po
4 4.0g/l,mnso4 10mg/l,vb
1 1.0mg/l,微量元素混合液1ml/l,谷氨酸1g/l,蛋氨酸0.2mg/l,α-酮戊二酸0.5g/l,每升流加葡萄糖中含4g氯化胆碱,4g甜菜碱,2mg v
b1
,苯酚红:需定容体积的2%,用naoh调ph至7.0-7.2,消泡剂1滴,其余为水;所述微量元素混合液组分含量为:钼酸铵0.28mg/l,mnso4·
h2o0.5mg/l,cuso4·
7h2o 0.5mg/l,上述成分称量固体后溶解于1l水中,在4℃保存。
[0155]
2.培养方法
[0156]
2.1斜面培养
[0157]
取-80℃保藏的实施例1所述用于产生羟脯氨酸的基因工程菌e.coli hyp划线接种于活化斜面,37℃培养12h,并传代一次。
[0158]
2.2种子培养
[0159]
用接种环刮取一环斜面种子接种于装有30ml种子培养基的500ml三角瓶中,九层纱布封口,36℃,200r/min培养10h。
[0160]
2.3发酵培养
[0161]
按15%接种量接种菌种活化后制备的种子液到装有发酵培养基的500ml三角瓶中(终体积为30ml),九层纱布封口,37℃,200r/min振荡培养,发酵过程中通过补加氨水维持ph在7.0-7.2;添加60%(m/v)葡萄糖溶液维持发酵进行(以苯酚红做指示剂,发酵液颜色不再变化时即视为缺糖,缺糖时补加1-2ml 60%(m/v)葡萄糖溶液)。发酵周期32h。
[0162]
摇瓶发酵32h后羟脯氨酸的产量可达40g/l。
[0163]
实施例3
[0164]
利用实施例1所述的大肠杆菌工程菌5l发酵罐发酵生产羟脯氨酸
[0165]
1.培养基
[0166]
1.1斜面培养基
[0167]
葡萄糖2g/l,蛋白胨10g/l,酵母浸出粉5g/l,氯化钠2.5g/l,kh2po
4 1.0g/l,mgso
4 0.2g/l,琼脂粉2.5%,用水溶解并定容至所需体积,用氢氧化钠调ph至7.0-7.2,121℃高压蒸汽锅灭菌20min后分装至试管和茄形瓶中。
[0168]
1.2种子培养基
[0169]
葡萄糖30g/l,酵母粉6g/l,蛋白胨4g/l,柠檬酸1g/l,(nh4)2so
4 3g/l,mgso4·
7h2o 0.4g/l,kh2po
4 1.5g/l,mnso
4 5mg/l,v
b1
0.5 mg/l,微量元素混合液(同实施例2)1ml/l,谷氨酸0.5g/l,蛋氨酸0.5mg/l,其余为水,在4℃保存。
[0170]
1.3发酵培养基
[0171]
葡萄糖20g/l,酵母粉6g/l,蛋白胨1g/l,柠檬酸1.5g/l,(nh4)2so
4 1g/l,mgso4
·
7h2o 1.5g/l,kh2po
4 4.0g/l,mnso
4 10mg/l,vb
1、3、5、12
各1.0mg/l,微量元素混合液(同实施例2)1ml/l,谷氨酸1g/l,蛋氨酸0.2mg/l,α-酮戊二酸0.5g/l,每升流加葡萄糖中含4g氯化胆碱,4g甜菜碱,2mg v
b1
,其余为水;上述成分称量固体后溶解于1l水中,在4℃保存。
[0172]
2.培养方法
[0173]
2.1斜面活化培养
[0174]
从-80℃冰箱保菌管中刮一环菌种,均匀涂布于活化斜面,37℃培养12h,转接茄形瓶继续扩大培养12h;
[0175]
2.2种子培养
[0176]
取适量无菌水于茄形瓶中,将菌悬液接入种子培养基中,ph维持在7.0-7.2,温度维持在36℃,溶氧在30%-60%之间,培养6h;
[0177]
2.3发酵培养
[0178]
等待种子菌体量od600至25左右,按照20%接种量接入新鲜的发酵培养基,开始发酵,发酵过程中控制ph稳定在7.0左右,温度维持在37℃,溶氧在30%-60%之间;当培养基中的葡萄糖消耗完之后,流加80%(m/v)的葡萄糖溶液,维持发酵培养基中的葡萄糖浓度在
0.1-1g/l;发酵周期45h;
[0179]
5l发酵罐发酵45h后反式-4-羟基脯氨酸的产量达到了129.2g/l,糖酸转化率为36.3%。发酵过程曲线见图16。
[0180]
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,本发明菌株的构建步骤不分先后顺序,本技术领域技术人员以本发明的方法或以本方法为基础进行的菌种改造等改进和润饰均视为本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1