一种乐伐替尼杂质及其制备方法与流程
1.本发明属于医药化工领域,具体涉及一种乐伐替尼杂质及其制备方法。
背景技术:
2.乐伐替尼(lenvatinib),化学名称为4-[3-氯-4-(环丙基氨基羰基)氨基苯氧基]-7-甲氧基-6-喹啉甲酰胺,分子式为c
21h19
cln4o4,cas登记号417716-92-8,结构式如下所示:
[0003][0004]
乐伐替尼是由日本卫材(eisai)公司研发的一种新型口服多靶点酪氨酸激酶抑制剂,该药被美国fda批准用于肝癌、甲状腺癌和晚期肾细胞癌治疗。2018年9月,乐伐替尼在中国获批上市,用于肝癌的靶向治疗,目前已是肝癌治疗使用最为广泛的一线药物。
[0005]
目前,已报道的乐伐替尼杂质有十几种,王国才、高飞飞等给出了乐伐替尼杂质a(4-(3-氯-4-(3-环丙基脲)苯氧基)-7-甲氧基喹啉-6-甲酸甲酯)和杂质b(4-(3-氯-4-(3-环丙基脲)苯氧基)-7-甲氧基喹啉-6-羧酸)的合成方法(王国才,高飞飞,卢娓.抗肿瘤药lenvatinib相关杂质的合成[j].广州化工.2019,47(09)),以及杂质4,4
’‑
[4,4
’‑
羰基双(脲二基)双(3-氯-4,1-次苯基)]双(氧)双(7-甲氧基喹啉-6-羧酰胺)的合成方法(高飞飞,高军龙,陈阿明.抗肿瘤药乐伐替尼相关杂质的合成[j].山东化工.2018,47(20))。而中国专利cn112409255a、cn114634446a、cn110117255a和cn108299294a均公开乐伐替尼相关杂质的合成方法,相关杂质的结构式分别如下所示:
[0006]
[0007][0008]
对乐伐替尼杂质以及合成方法进行研究,对乐伐替尼的质量控制起到非常重要的作用。基于此,本技术开发了一种新的乐伐替尼杂质及其制备方法。
技术实现要素:
[0009]
针对上述问题,本发明提供了一种乐伐替尼杂质及其制备方法。本发明首先在乐伐替尼制备过程中发现了一种新的乐伐替尼杂质,然后提供了一种可以稳定、高产率及高纯度地合成该杂质的方法,该杂质可作为杂质对照品对乐伐替尼的制备过程进行严格、精确的质量监控。
[0010]
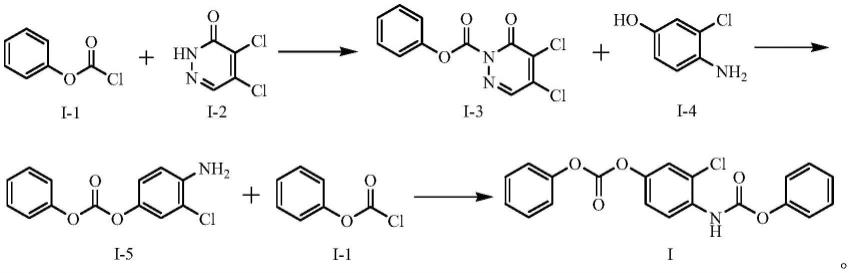
本发明首先提供了一种乐伐替尼杂质,该杂质主要在氯甲酸苯酯与4-氨基-3-氯苯酚反应制备乐伐替尼中间体过程中产生(合成路线参见ep1683785a1、cn101337930a等专利),其结构式如下ⅰ所示,化合物名称:苯基(2-氯-4-(苯氧羰基)氧基)苯基)氨基甲酸酯。
[0011][0012]
本发明还提供了上述乐伐替尼杂质的制备方法,其特征是,包括以下步骤:
[0013]
1)在缚酸剂作用下,氯甲酸苯酯(式i-1化合物)与4,5-二氯哒嗪-3(2h)-酮(式i-2化合物)反应,得到式i-3所示中间体;
[0014]
2)在碱催化下,式i-3所示中间体与4-氨基-3-氯苯酚反应,得到式i-5所示中间体;
[0015]
3)在缚酸剂作用下,式i-5所示中间体与氯甲酸苯酯进行反应,得到乐伐替尼杂质ⅰ。
[0016]
合成路线如下所示。
[0017][0018]
由于氯甲酸苯酯无法直接与4-氨基-3-氯苯酚的酚羟基反应,本技术以4,5-二氯哒嗪-3(2h)-酮作为过渡,制备出中间体i-5,然后再与氯甲酸苯酯反应,得到最终产物。
[0019]
进一步地,步骤1)中反应溶剂为二氯甲烷、氯仿、甲苯、乙腈、甲醇和乙醇中的一种或几种,优选为二氯甲烷。
[0020]
进一步地,步骤1)中,反应温度为-10~20℃,优选反应温度为-5~5℃。
[0021]
进一步地,步骤1)和3)中所用缚酸剂为三乙胺、吡啶、dmap、碳酸钾,优选使用三乙胺、吡啶;进一步地,氯甲酸苯酯(式i-5所示中间体)与缚酸剂的投料摩尔比为1:1~1.5,优选为1:1.3。
[0022]
进一步地,步骤1)中氯甲酸苯酯与4,5-二氯哒嗪-3(2h)-酮的投料摩尔比为1:1~1.5,优选为1:1.25。
[0023]
进一步地,步骤2)中反应溶剂为甲苯、dmf、dmso和四氢呋喃中的一种或几种,优选为甲苯。
[0024]
进一步地,步骤2)中所用碱催化剂为叔丁醇钾、氢氧化钠、氢氧化钾、碳酸钾等,优选使用叔丁醇钾;进一步地,式i-3中间体与叔丁醇钾投料比为1:1~1.5,优选为1:1.2。
[0025]
进一步地,步骤2)中,反应温度为10~40℃,优选反应温度为20~30℃。
[0026]
进一步地,步骤2)中式i-3中间体与4-氨基-3-氯苯酚的投料摩尔比为1:1~1.5,优选为1:1.2。
[0027]
进一步地,步骤3)中反应溶剂包括甲苯、dmf、dmso和四氢呋喃中的一种或几种,优选为dmf。
[0028]
进一步地,步骤3)中,反应温度为10~40℃,优选反应温度为20~30℃。
[0029]
进一步地,步骤3)中式i-5所示中间体与氯甲酸苯酯投料摩尔比为1:1~1.5,优选为1:1.2。
[0030]
优选的,其合成方法具体包括以下步骤:
[0031]
1)将4,5-二氯哒嗪-3(2h)-酮、三乙胺和二氯甲烷加入反应容器中,加入氯甲酸苯酯,加毕,-5~5℃保温反应0.5~1.0h,反应完毕,盐洗,有机相干燥,减压浓缩至干,得到式i-3所示中间体;
[0032]
2)将式i-3所示中间体、4-氨基-3-氯苯酚、叔丁醇钾和甲苯加入反应容器中,加毕,室温搅拌过夜,反应完毕,加入二氯甲烷和氢氧化钠水溶液,搅拌分液,有机相洗涤、干燥,浓缩至干,得到式i-5所示中间体;
[0033]
3)将式i-5所示中间体、吡啶和dmf加入反应容器中,加毕,控温<25℃,缓慢加入氯甲酸苯酯,加毕,室温反应2~5h,反应完毕,加入纯化水,加酸调节ph=4~5,乙酸乙酯萃取,有机相盐洗、干燥,减压浓缩至干,得到乐伐替尼杂质。
[0034]
本发明的技术效果是:该杂质的制备,可作为杂质对照品对乐伐替尼的制备过程进行严格、精确的质量监控。该制备方法可以稳定、高产率及高纯度地合成乐伐替尼杂质。
附图说明
[0035]
图1为本发明的乐伐替尼杂质的核磁共振氢谱谱图;
[0036]
图2为本发明的乐伐替尼杂质的核磁共振碳谱谱图;
具体实施方式
[0037]
下面结合具体实施例对本发明进行进一步说明,但本发明所申请保护的内容和范围并不受下述实施例的限制。
[0038]
实施例1
[0039]
1)杂质中间体i-3的制备
[0040][0041]
将4,5-二氯哒嗪-3(2h)-酮13.2g、三乙胺8.4g和二氯甲烷100ml依次加入干燥反应瓶中,并降温至0℃,缓慢加入氯甲酸苯酯10.0g,加毕,保温反应0.5h,反应完毕,加入二氯甲烷300ml将其稀释,饱和氯化钠溶液洗涤两次,有机相用无水硫酸钠干燥,过滤,滤液减压浓缩至干,得杂质中间体i-3 16.8g,收率92%,hplc纯度98.5%。
[0042]
2)杂质中间体i-5的制备
[0043][0044]
将杂质中间体i-3 16.0g、4-氨基-3-氯苯酚9.7g、叔丁醇钾7.6g和甲苯100ml依次加入干燥反应瓶中,加毕,室温搅拌过夜,反应完毕,加入二氯甲烷100ml和100ml 25%氢氧化钠水溶液,搅拌分液,有机相用纯水洗涤,无水硫酸钠干燥,过滤,滤液减压浓缩至干,得杂质中间体i-5 11.5g,收率78%,hplc纯度97.5%。
[0045]
3)乐伐替尼杂质i的制备
[0046][0047]
将杂质中间体i-5 11.0g、吡啶4.3g和dmf 60ml依次加入干燥反应瓶中,加毕,控温<25℃,缓慢加入氯甲酸苯酯7.8g,加毕,室温反应3h,反应完毕,加入纯化水60ml,盐酸调节ph=4~5,计时搅拌1h,乙酸乙酯萃取,有机相用饱和氯化钠洗涤,无水硫酸钠干燥,过滤,滤液减压浓缩至干,得乐伐替尼杂质i12.8g,收率80%,hplc纯度98.3%。
[0048]
产物的核磁共振氢谱谱图如图1所示,核磁共振碳谱谱图如图2所示。
[0049]
实施例2
[0050]
1)杂质中间体i-3的制备
[0051]
将4,5-二氯哒嗪-3(2h)-酮20.6g、三乙胺13.0g和二氯甲烷150ml依次加入干燥反应瓶中,并降温至0℃,缓慢加入氯甲酸苯酯15.6g,加毕,保温反应40min,反应完毕,加入二氯甲烷500ml将其稀释,饱和氯化钠溶液洗涤两次,有机相用无水硫酸钠干燥,过滤,滤液减压浓缩至干,得杂质中间体i-3 26.1g,收率91.8%,hplc纯度98.6%。
[0052]
2)杂质中间体i-5的制备
[0053]
将杂质中间体i-3 22.7g、4-氨基-3-氯苯酚13.8g、叔丁醇钾10.7g和甲苯150ml依次加入干燥反应瓶中,加毕,室温搅拌过夜,反应完毕,加入二氯甲烷150ml和150ml 25%氢氧化钠水溶液,搅拌分液,有机相用纯水洗涤,无水硫酸钠干燥,过滤,滤液减压浓缩至干,得杂质中间体i-5 16.8g,收率79.8%,hplc纯度97.8%。
[0054]
3)乐伐替尼杂质i的制备
[0055]
将杂质中间体i-5 13.1g、吡啶5.1g和dmf 70ml依次加入干燥反应瓶中,加毕,控温<25℃,缓慢加入氯甲酸苯酯9.4g,加毕,室温反应2.5h,反应完毕,加入纯化水70ml,盐酸调节ph=4~5,计时搅拌1h,乙酸乙酯萃取,有机相用饱和氯化钠洗涤,无水硫酸钠干燥,过滤,滤液减压浓缩至干,得乐伐替尼杂质i15.5g,收率81%,hplc纯度98.5%。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1