氟氧头孢酸杂质的制备方法与流程
本发明属于精细有机合成领域,具体涉及氟氧头孢酸杂质的制备方法。
背景技术:
1、氟氧头孢钠由日本盐野义制药株式会社开发,为白色至浅黄白色轻质粉末或块状物。氟氧头孢钠是一种广谱的氧头孢烯(氧杂头孢菌素)抗菌药物,对β-内酰胺酶十分稳定,几乎不产生耐药性,而且肾毒性很低,因此氧氟头孢钠的合成具有重要的临床和工业价值。氟氧头孢钠于2019年11月28日以商品名“氟吗宁”批准在中国上市,制剂规格为每瓶含0.5g、1.0g氟氧头孢。
2、氟氧头孢钠化学名:7-二氟甲基硫代乙酰胺基-7a-甲氧基-3-[1(2-羟乙基-1h-四氮唑-5-疏甲基)]-1-去硫-1-氧杂-3-头孢-4-羧酸钠
3、化学结构式:
4、
5、杂质的制备是原料药和制剂质量研究的重要组成部分。影响药品使用安全的主要因素就是杂质的控制和研究,为了确保药品安全性,杂质必须充分研究。原研针对其生产工艺所制备出来的原料药已经进行了充分的临床研究,仿制药不进行临床试验,由于可能采用不同的合成工艺,杂质谱也有可能不同,可能某些未被关注的高毒性杂质会导致严重后果。而氢溴酸伏硫西汀杂质种类繁多,杂质情况复杂,不易购买,且售价昂贵。因此对一些关键的杂质的制备和研究是非常必要的。
6、氟氧头孢酸原料在生产过程中有产生杂质(6r,7r)-甲基-7-(2-(二氟甲氧基硫代)乙酰胺基)-3-(((1-(2-羟乙基)-1h-四唑-5-基)硫烷基)甲基)-7-甲氧基-8-氧代-5-氧杂-1-氮杂双环[4.2.0]辛-2-烯-2-羧酸酯,其化学结构式如下:
7、
8、该杂质属于工艺杂质。因此对该杂质的控制对氟氧头孢酸产品质量至关重要,而且其制备方法尚未有文献报道,因此该杂质的制备,精制及进行后续的结构确证对有效的控制氟氧头孢酸原料的质量有着重要的意义。
技术实现思路
1、本发明目的在于提供氟氧头孢酸杂质的制备方法。该杂质可以作为氟氧头孢酸质量控制参照标准品的应用。
2、为实现上述目的,本发明采用如下技术方案:
3、氟氧头孢酸杂质的制备方法,包括以下步骤:
4、一.通过甲氧基化合成反应及甲氧基化分离制备中间体1
5、(1)甲氧基化合成反应
6、1、在10~20重量份的有机溶剂中、加入1重量份的氟氧头孢母核,氮气保护下搅拌,使物料完全溶解,得到料液;
7、2、在氮气保护下将料液冷却到-60℃以下,控温-50~-60℃,滴加0.2~0.5重量份的次氯酸叔丁酯,反应40~45min;
8、3、控温-55~-60℃,滴加2~4重量份的10%甲醇锂甲醇溶液,反应1~1.1h;
9、4、反应完毕加入亚硫酸钠和醋酸淬灭反应;
10、5、加入饱和食盐水萃取分层,有机相浓缩至干,得到残留液1(有机层浓缩后的油状物);
11、6、将残留液1加入甲醇和有机弱酸后,25℃搅拌2~3h,冷却至0±1℃,继续搅拌10~12小时,过滤除去固体后,滤液浓缩至干,得残留液2;
12、(2)甲氧基化分离制备(中间体1)
13、采用正相硅胶柱(硅胶为300-400目)、干法上样方式,对残留液2进行如下处理:在上述残留液2中加入2~3重量份甲醇与5~6重量份的二氯甲烷完全溶解,100-200目硅胶拌样,然后先后用c:m(二氯甲烷:甲醇)=100:1、50:1的洗脱液,洗脱约2个柱体积后,继续用c:m(二氯甲烷:甲醇)=30:1的洗脱液洗脱约2个柱体积。用tlc点板确认大部分的中间体1洗脱下来后,改成c:m(二氯甲烷:甲醇)=10:1的洗脱液,洗脱约2个柱体积,最后用c:m(二氯甲烷:甲醇)=1:1的洗脱液完成剩余中间体1的收集;
14、二.通过脱苯甲酰基保护获得中间体2反应液
15、1、在2~3重量份的二氯甲烷中、加入0.5重量份的五氯化磷,氮气保护下滴加0.2~0.3重量份的有机碱,于25℃搅拌15~20分钟;
16、2、于25℃滴加中间体1的二氯甲烷溶液(1重量份的中间体1溶解在7~8重量份的二氯甲烷中);
17、3、反应液变澄清后升温至40~42℃,于40~42℃反应60~65分钟;
18、4、在氮气保护下冷却到0℃并滴加10~15重量份的甲醇,于0±1℃反应2~2.1小时(液相检测原料<1%);
19、5、反应完毕,将反应液滴加到10~15重量份的碳酸氢钠水溶液中,同时用碳酸钠水溶液调节体系ph值至5.5;
20、6、萃取分层,将有机层干燥除水后得到中间体2反应液;
21、三.通过7位酰化反应获得中间体3反应液
22、1、在2~3重量份的二氯甲烷中,加入0.4~0.5重量份的二氟甲基硫代乙酸钾和2~3重量份的纯化水,于0℃搅拌,滴加10%无机酸调节ph至1.0,分层,有机层用饱和氯化钠洗涤,无水硫酸镁干燥20~25分钟,过滤,得到二氟甲基硫代乙酸的二氯甲烷溶液;
23、2、加入1~2重量份的有机碱,降温至-20℃以下,并于-20℃以下加入中间体2反应液,搅拌10~15分钟;
24、3、降温至-47℃以下,快速加入0.8~1.2重量份的三氯氧磷;
25、4、升温至-15℃反应20分钟,hplc检测原料反应完全;
26、5、反应完毕,将反应液加入到0℃下的3~4重量份的水和2~3重量份的二氯甲烷的混合溶液中;
27、6、萃取分层,将有机层干燥除水后得到中间体3反应液;
28、四.通过3位硫化反应获得氟氧头孢酸杂质
29、1、反应瓶中加入0.8~1.2重量份的1-(2-羟乙基)-1h-四氮唑-5-硫钠和2~3重量份的水,及0.1~0.2重量份的tbab,搅拌均匀;
30、2、于20℃以下加入中间体3反应液,升温至25℃,加入0.5~1.2重量份30%盐酸,保温反应150分钟,其间用饱和碳酸氢钠水溶液调ph值至6.0,hplc检测产物含量<2%;
31、3、静置分层,分出有机层,有机层浓缩至干,获得残留液3;
32、4、将残留液3(有机层浓缩后的油状物)加入脂肪族醇和水的混合液,冷却至0±1℃,搅拌析晶10小时;
33、5、抽滤得到白色固体,减压干燥,获得氟氧头孢酸杂质,即(6r,7r)-甲基-7-(2-(二氟甲氧基硫代)乙酰胺基)-3-(((1-(2-羟乙基)-1h-四唑-5-基)硫烷基)甲基)-7-甲氧基-8-氧代-5-氧杂-1-氮杂双环[4.2.0]辛-2-烯-2-羧酸酯,其结构式如下:
34、
35、本发明中,所述有机溶剂为二氯甲烷或氯仿。
36、所述有机弱酸为甲酸、冰醋酸或草酸。
37、所述有机碱为吡啶、2,6-二甲基吡啶、喹啉或三乙胺。
38、所述无机酸为盐酸、硫酸或磷酸。
39、所述脂肪族醇为乙醇、甲醇、异丙醇、正丁醇或异戊醇。
40、中间体1产生机理分析如下:
41、
42、目标产物合成路线如下:
43、
44、本发明以氟氧头孢母核为起始原料,经甲氧基化、脱苯甲酰基保护、7位酰化反应、3位硫化反应制得(6r,7r)-甲基-7-(2-(二氟甲氧基硫代)乙酰胺基)-3-(((1-(2-羟乙基)-1h-四唑-5-基)硫烷基)甲基)-7-甲氧基-8-氧代-5-氧杂-1-氮杂双环[4.2.0]辛-2-烯-2-羧酸酯产品。本发明具有以下优点:
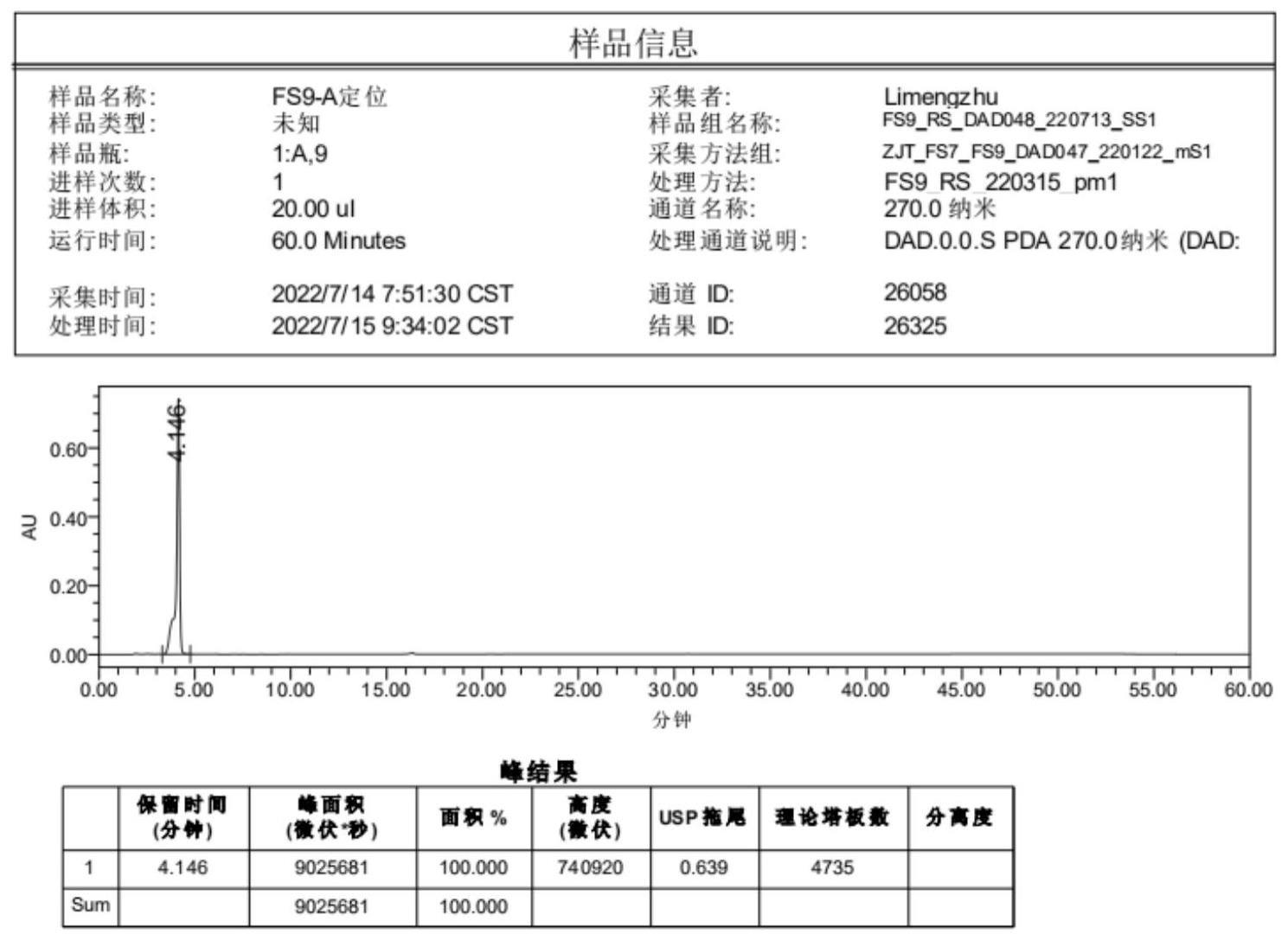
45、1、本发明所制备的杂质可以作为氟氧头孢酸的杂质对照品,主要是用于对氟氧头孢酸的关键中间体氟氧头孢酯进行杂质定位,为后续的氟氧头孢酸的杂质研究提供基础。
46、2、本发明同时对氟氧头孢杂质杂质进行了结构确证,进行了质谱和核磁共振的检测,对氟氧头孢原料药的质量研究至关重要。
47、3、本发明采用一锅法制备,操作工艺简捷,合成成本低,产品纯度高,可达99%以上。
- 还没有人留言评论。精彩留言会获得点赞!