一种抗真菌药物的合成方法与流程
本发明涉及药物合成,具体涉及一种抗真菌药物的合成方法,特别是涉及一种1-{(2r,3r)-3-[4-(4-氰基苯基)-1,3-噻唑-2-基]-2-(2,5-二氟苯基)-2-羟基丁基}-4-[(1rs)-1-({甲基[3-({[(甲基氨基)乙酰基]氧基}甲基)吡啶-2-基]氨基甲酰基}氧基)乙基]-1h-1,2,4-三唑-4-鎓单硫酸盐的合成方法。
背景技术:
1、1-{(2r,3r)-3-[4-(4-氰基苯基)-1,3-噻唑-2-基]-2-(2,5-二氟苯基)-2-羟基丁基}-4-[(1rs)-1-({甲基[3-({[(甲基氨基)乙酰基]氧基}甲基)吡啶-2-基]氨基甲酰基}氧基)乙基]-1h-1,2,4-三唑-4-鎓单硫酸盐是由安斯泰来(astellas)和巴塞利亚(basilea)联合开发的用于侵袭性曲霉菌感染和侵袭性毛霉菌感染治疗的前体药物,于2015年3月获得fda批准上市。
2、1-{(2r,3r)-3-[4-(4-氰基苯基)-1,3-噻唑-2-基]-2-(2,5-二氟苯基)-2-羟基丁基}-4-[(1rs)-1-({甲基[3-({[(甲基氨基)乙酰基]氧基}甲基)吡啶-2-基]氨基甲酰基}氧基)乙基]-1h-1,2,4-三唑-4-鎓单硫酸盐属于唑类抗真菌药物,是4-(2-((2r,3r)-3-(2,5-二氟苯基)-3-羟基-4-(1h-1,2,4-三唑-1-基)丁烷-2-基)噻唑-4-基)苯腈的前药形式,与其他的三氮唑抗菌药相比,具有很强的水溶性,因此不需要在其制剂中加入β-环糊精以提高溶解度,消除了环糊精加入造成的肾脏毒性的可能,同时其口服制剂的吸收不受食物和胃酸影响,有较高的生物利用度。1-{(2r,3r)-3-[4-(4-氰基苯基)-1,3-噻唑-2-基]-2-(2,5-二氟苯基)-2-羟基丁基}-4-[(1rs)-1-({甲基[3-({[(甲基氨基)乙酰基]氧基}甲基)吡啶-2-基]氨基甲酰基}氧基)乙基]-1h-1,2,4-三唑-4-鎓单硫酸盐的代谢毒性低,口服该药物的尿溶度可忽略不计,静脉给药后尿浓度仅略微提高,对于肾功能减退的患者可正常使用。此外,1-{(2r,3r)-3-[4-(4-氰基苯基)-1,3-噻唑-2-基]-2-(2,5-二氟苯基)-2-羟基丁基}-4-[(1rs)-1-({甲基[3-({[(甲基氨基)乙酰基]氧基}甲基)吡啶-2-基]氨基甲酰基}氧基)乙基]-1h-1,2,4-三唑-4-鎓单硫酸盐具有良好的安全性和耐受性,常见的副作用为头痛和轻度胃肠道症状,该药物造成肝脏的毒性是可逆的,停止用药即可恢复。
3、由于1-{(2r,3r)-3-[4-(4-氰基苯基)-1,3-噻唑-2-基]-2-(2,5-二氟苯基)-2-羟基丁基}-4-[(1rs)-1-({甲基[3-({[(甲基氨基)乙酰基]氧基}甲基)吡啶-2-基]氨基甲酰基}氧基)乙基]-1h-1,2,4-三唑-4-鎓具有热不稳定性导致重结晶纯化困难、结构中含有多个成盐位点导致硫酸根个数难控制,因此实现由1-{(2r,3r)-3-[4-(4-氰基苯基)-1,3-噻唑-2-基]-2-(2,5-二氟苯基)-2-羟基丁基}-4-[(1rs)-1-({甲基[3-({[(甲基氨基)乙酰基]氧基}甲基)吡啶-2-基]氨基甲酰基}氧基)乙基]-1h-1,2,4-三唑-4-鎓季铵盐的卤素转变为硫酸氢盐的盐型转化是工艺的技术难点,现有报道的1-{(2r,3r)-3-[4-(4-氰基苯基)-1,3-噻唑-2-基]-2-(2,5-二氟苯基)-2-羟基丁基}-4-[(1rs)-1-({甲基[3-({[(甲基氨基)乙酰基]氧基}甲基)吡啶-2-基]氨基甲酰基}氧基)乙基]-1h-1,2,4-三唑-4-鎓单硫酸盐合成或纯化工艺中,大都是针对上述问题进行优化,例如中国专利申请cn106467534a报道了1-{(2r,3r)-3-[4-(4-氰基苯基)-1,3-噻唑-2-基]-2-(2,5-二氟苯基)-2-羟基丁基}-4-[(1rs)-1-({甲基[3-({[(甲基氨基)乙酰基]氧基}甲基)吡啶-2-基]氨基甲酰基}氧基)乙基]-1h-1,2,4-三唑-4-鎓单硫酸盐的纯化方法、cn106916152a报道了通过氧化还原反应精确控制硫酸根个数的方法等。
4、而对于保护基boc的脱保护,则主要还是沿用原研us7459561b2报道的将式ii的化合物经hcl溶液脱boc得到式iv化合物的工艺,如下所示:
5、
6、原研us7459561b2的工艺存在两个问题,其一是式iv化合物为盐酸盐形式,极易吸潮,需要严格控制实验环境的水分,为操作带来诸多不便,增加控制过程成本;其二是由于hcl酸性强,在反应过程中会导致工艺杂质1-((2r,3r)-3-(4-(4-(叔丁氧基(亚胺基)甲基)苯基)噻唑-2-基)-2-(2,5-二氟苯基)-2-羟基丁基)-4-(1-((甲基(3-(((甲基甘氨基)氧基)甲基)吡啶-2-基)氨甲酰)氧基)乙基)-1h-1,2,4-三唑-4-鎓(式v的化合物)的生成,该杂质为式ii化合物脱boc时产生的叔丁基离子与1-{(2r,3r)-3-[4-(4-氰基苯基)-1,3-噻唑-2-基]-2-(2,5-二氟苯基)-2-羟基丁基}-4-[(1rs)-1-({甲基[3-({[(甲基氨基)乙酰基]氧基}甲基)吡啶-2-基]氨基甲酰基}氧基)乙基]-1h-1,2,4-三唑-4-鎓反应生成,难以通过简单纯化去除,需要通过柱层析或制备纯化。
7、
8、综上所述,迫切需要开发一种新的式ii化合物脱boc时能够避免易吸潮中间体式iv化合物并控制副产物式v化合物生成的操作简便、易于工业化的1-{(2r,3r)-3-[4-(4-氰基苯基)-1,3-噻唑-2-基]-2-(2,5-二氟苯基)-2-羟基丁基}-4-[(1rs)-1-({甲基[3-({[(甲基氨基)乙酰基]氧基}甲基)吡啶-2-基]氨基甲酰基}氧基)乙基]-1h-1,2,4-三唑-4-鎓单硫酸盐的合成方法。
技术实现思路
1、本发明的目的是提供一种1-{(2r,3r)-3-[4-(4-氰基苯基)-1,3-噻唑-2-基]-2-(2,5-二氟苯基)-2-羟基丁基}-4-[(1rs)-1-({甲基[3-({[(甲基氨基)乙酰基]氧基}甲基)吡啶-2-基]氨基甲酰基}氧基)乙基]-1h-1,2,4-三唑-4-鎓单硫酸盐的合成方法,包括将式ii的化合物在三氟乙酸和六氟异丙醇下脱boc得到式iii的化合物,式iii的化合物转盐得到式i的化合物。本发明的合成方法,避免了现有工艺采用hcl脱boc得到的式iv易吸潮降解,而且减少了现有工艺采用hcl、h2so4、tfa与其他溶剂脱boc时副产物式v的产生,无需纯化就可将该杂质控制在0.10%以下,工艺操作简单,总纯度可达99%以上,各项指标合格。
2、本发明的技术方案如下:
3、一种1-{(2r,3r)-3-[4-(4-氰基苯基)-1,3-噻唑-2-基]-2-(2,5-二氟苯基)-2-羟基丁基}-4-[(1rs)-1-({甲基[3-({[(甲基氨基)乙酰基]氧基}甲基)吡啶-2-基]氨基甲酰基}氧基)乙基]-1h-1,2,4-三唑-4-鎓单硫酸盐的合成方法,该方法包括如下步骤:
4、(1)将式ii的化合物在三氟乙酸的存在下在反应溶剂中脱boc生成式iii的化合物:
5、以及
6、(2)向步骤(1)得到的式iii的化合物中添加硫酸转化为式i的硫酸盐化合物:
7、
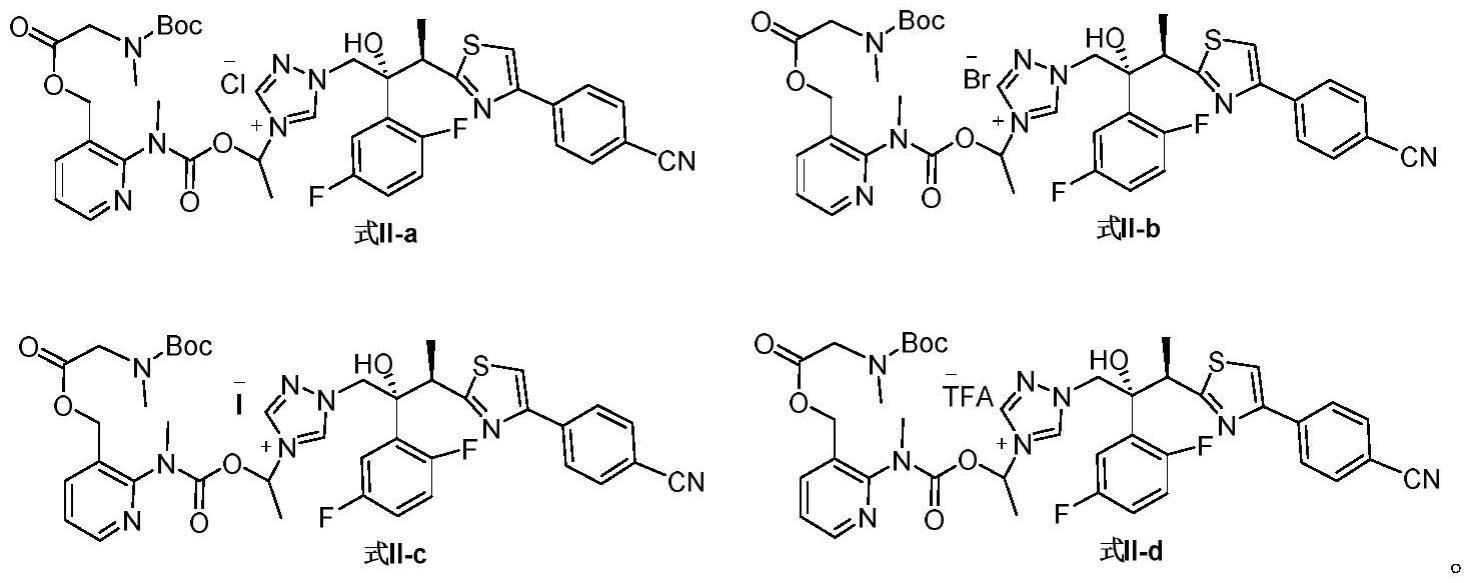
8、其中,x表示卤素或tfa。
9、根据本发明,式ii的化合物选自如下:
10、
11、根据本发明,步骤(1)中所用的原料式ii化合物为直接购买或根据已有文献报道合成。
12、根据本发明,在步骤(1)中,所述反应溶剂为非叔丁基离子的反应溶剂,例如为六氟异丙醇。
13、根据本发明,在步骤(1)中,反应温度为0~60℃,优选为40~45℃。
14、根据本发明,在步骤(1)中,所述式ii的化合物与所述三氟乙酸之间的摩尔投料比为1:0.8~1:40,优选为1:3~1:5(例如为1mmol:0.8mmol~1mmol:40mmol,优选为1mmol:3mmol~1mmol:5mmol)。
15、根据本发明,步骤(1)中,所述式ii的化合物与所述反应溶剂的质量体积比为1:1~1:100,优选为1:5~1:15(例如为1g:1ml~1g:100ml,优选为1g:5ml~1g:15ml)。
16、根据本发明,在步骤(2)中,向步骤(1)得到的式iii的化合物中添加硫酸,析出固体,过滤,滤饼用水溶解,加入氢氧化钡中和过量硫酸,过滤,水相冻干,以得到式i的硫酸盐化合物。
17、与现有技术相比,本发明的优势至少包括以下方面:
18、本发明采用了特殊的溶剂与三氟乙酸相配合,副产物式v化合物在0.10%以下,无需通过柱层析或制备纯化去除该杂质,适宜工业化放大生产。
19、反应完全后加入硫酸析出的4-(2-((2r,3r)-3-(2,5-二氟苯基)-3-羟基-4-(1h-1,2,4-三唑-1-基)丁烷-2-基)噻唑-4-基)苯腈多硫酸盐不易吸潮,稳定性优于4-(2-((2r,3r)-3-(2,5-二氟苯基)-3-羟基-4-(1h-1,2,4-三唑-1-基)丁烷-2-基)噻唑-4-基)苯腈盐酸盐,无需在氮气环境中过滤,操作简单。
- 还没有人留言评论。精彩留言会获得点赞!