一种高效构建多基因改造酵母菌的方法及其应用与流程
本发明属于基因工程。更具体地,涉及一种高效构建多基因改造酵母菌的方法及其应用。
背景技术:
1、槐糖脂是一类具有常规表面活性剂所具有的增溶、乳化、润湿、发泡、分散和降低表面张力等通用性能,且对环境的耐受性极强的生物表面活性剂。熊蜂生假丝酵母菌(starmerella bombicola,s.bombicola)是一种能够合成槐糖脂的非致病假丝酵母菌。在现有已知的产槐糖脂的菌株中,熊蜂生假丝酵母菌的产量最高,有着最好的应用前景。但和常用化学表面活性剂相比,目前利用熊蜂生假丝酵母菌发酵生成槐糖脂的成本仍然较高。因此,有必要提供高产槐糖脂的熊蜂生假丝酵母菌菌株或可在较短时间内发酵生成槐糖脂的熊蜂生假丝酵母菌菌株,以便从源头上降低利用熊蜂生假丝酵母菌发酵生成槐糖脂的生产成本。
2、现虽已有通过敲除熊蜂生假丝酵母菌的特定基因来提高其槐糖脂产量的报道,但可在更短时间内发酵生成等量槐糖脂的熊蜂生假丝酵母菌菌株的报道较少。且有研究发现,熊蜂生假丝酵母菌中槐糖脂的合成路径受多个关键酶基因的调控,同时还受到脂肪酸的β-氧化、葡萄糖分解等多个代谢路径的影响。为进一步提高熊蜂生假丝酵母菌产槐糖脂的能力,可能需要对不同通路的多个关键酶基因进行改造,同时还需引入便于筛选阳性克隆的抗性基因,这就对长片段dna载体的构建效率和转化效率提出了非常高的技术要求。
3、现有对熊蜂生假丝酵母菌进行多基因改造的方法主要为逐步改造,即先对熊蜂生假丝酵母菌的单个基因通过同源重组的方式进行敲除或过表达,筛选出改造成功的阳性菌株后,再以该菌株作为出发菌株再进行下一个基因的改造,该方法存在效率低、耗时长、操作繁杂和成本高的问题。此外,目前虽有利于融合pcr获得线性敲除盒并转化酵母菌对其进行基因敲除的方法,但该方法构建的线性敲除盒仅含单个基因,整体长度也仅为2729bp,该方法不一定适用于多基因长片段dna载体的构建和转化。在对熊蜂生假丝酵母菌合成槐糖脂相关代谢通路中的关键酶进行基因改造时,简单、快速的多基因片段载体的构建及高效的转化效率,是加快高产槐糖脂稳定菌株开发的重要保障。因此,亟需提供一种高效构建多基因改造酵母菌的方法。
技术实现思路
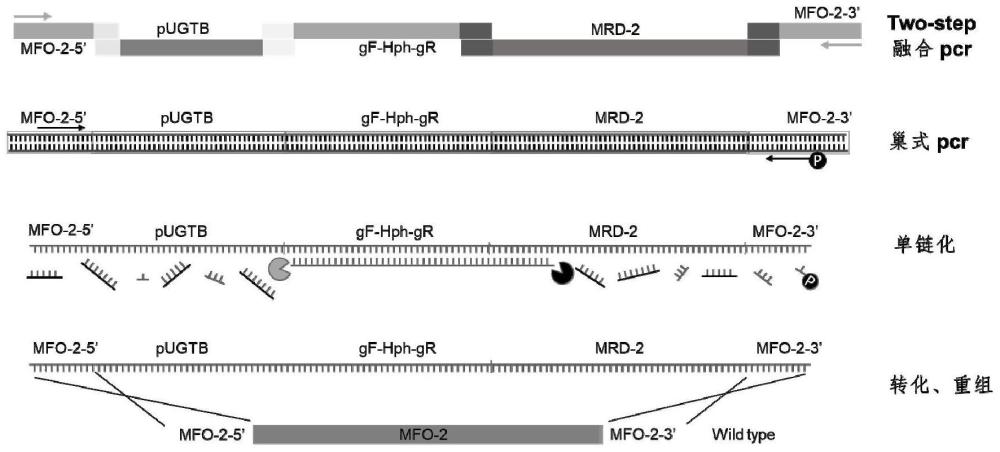
1、本发明针对上述现有技术的不足,提供了一种高效构建多基因改造酵母菌的方法,并利用此方法构建得到了一种能在更短时间能发酵生成等量槐糖脂的多基因改造的熊蜂生假丝酵母菌菌株。
2、本发明的目的是提供一种高效构建多基因改造酵母菌的方法。
3、本发明的另一目的是提供一种利用所述方法构建的多基因改造的酵母菌。
4、本发明上述目的通过以下技术方案实现:
5、现有的对酵母菌进行多基因改造的方法主要为逐步改造,此类方法存在效率低、耗时长、操作繁杂和成本高等问题。虽通过融合pcr可以将多个基因连接为一条长双链dna,但若待融合的基因片段中包含较长的dna序列以及基因片段数量较多时,则难以通过常规融合pcr反应进行融合连接。此外,长度大于8000bp的双链载体难以通过常规电转方法转入酵母菌中,不利于酵母菌的多基因改造。为克服上述技术问题,本发明提供了一种高效构建多基因改造酵母菌的方法,所述方法包括以下步骤:
6、s1.确定待敲除基因、一个或若干个待过表达基因;以待敲除基因的上游同源臂作为第一个dna片段,以待敲除基因的下游同源臂作为最后一个dna片段,所述一个或若干个待过表达基因为依次连接在所述第一个dna片段和最后一个dna片段中间的dna片段;
7、s2.制备s1所述的所有dna片段,通过两步法融合pcr和巢氏pcr将所有dna片段连接成用于多基因改造的长双链dna目标载体;
8、其中,所述两步法融合pcr的反应程序中的延伸时间以酶反应速率为30~60sec/kb计算;
9、s3.将s2所得长双链dna目标载体单链化为长单链dna目标载体并转化酵母菌感受态细胞,筛选阳性转化子。
10、具体地,s1所述待过表达的基因包括其编码区、启动子和终止子的核苷酸序列。
11、具体地,s1所述第一个dna片段的3’端引入了与其待连接dna片段的5’端同源的长为35~45bp的序列,最后一个dna片段的5’端引入了与其待连接dna片段的3’端同源的长为35~45bp的序列,其余中间dna片段的5’端引入了与其连接的dna片段的3’端同源的长为35~45bp的序列,3’端引入了与其连接的dna片段的5’端同源的长为35~45bp的序列。
12、为便于多基因改造酵母菌的筛选,所述待过表达的基因还包括抗性基因。所述抗性基因的dna片段的5’端引入了与其连接的dna片段的3’端同源的长为35~45bp的序列,3’端引入了与其连接的dna片段的5’端同源的长为35~45bp的序列。
13、可选地,可通过人工合成或pcr方法制备得到所述的所有dna片段。
14、具体地,s2所述的两步法融合pcr包括第一步无引物融合pcr和第二步有引物融合pcr;第一步无引物融合pcr的反应体系包括所有dna片段以及高保真酶,反应程序中的延伸时间以酶反应速率为30~60sec/kb计算;第二步有引物融合pcr以第一步无引物融合pcr的反应产物为模板,利用待敲除基因的上游同源臂的上游引物以及待敲除基因的下游同源臂的下游引物进行pcr扩增反应,反应程序中的延伸时间以酶反应速率为30~60sec/kb计算。
15、更具体地,第一步无引物融合pcr的反应体系为:将制备得到的带有同源序列的各dna片段以相同的摩尔数加入primestar max premix反应体系中,不添加任何引物,进行5~10个循环,延伸速度为30~60s/kb。
16、优选地,第一步无引物融合pcr进行10个循环。
17、具体地,当拟构建的长双链dna目标载体的长度超过5000bp时,添加的dna片段的总量需大于500ng。
18、具体地,s2所述的第二步有引物融合pcr进行25~30个循环。
19、具体地,s2所述巢氏pcr仅包括一对引物,其中一条引物的5’端需进行磷酸化修饰。
20、具体地,s3所述将长双链dna目标载体单链化为长单链dna目标载体包括以下步骤:
21、s31.向s2所得产物中加入dna解旋酶a进行识别长双链dna目标载体中5’端被磷酸化修饰的链并初始消化该链的反应;
22、s32.向s31的反应液中加入dna解旋酶b,完全消化5’端被磷酸化修饰的链。
23、具体地,步骤s31中,每10~15μg产物加入5μl dna解旋酶a和10μl反应buffer。
24、具体地,s31与s32的反应条件相同,反应条件为37℃反应5~15min,80℃反应5min,4℃反应10min。
25、优选地,37℃反应时间为10min。
26、当所构建的长双链dna目标载体的长度低于10000bp时均可采用上述反应体系和条件。若长于10000bp,则需适当调整反应体系和条件,延长反应时间。
27、具体地,s3在电转化酵母菌感受态细胞时,反应液中需加入终浓度为0.1~1%(v/v)的dmso(二甲基亚砜)。
28、优选地,加入终浓度为0.5%的dmso。
29、具体地,电转条件为施加5ms和2.5kv的脉冲。
30、鉴于利用本发明所述方法可以实现对酵母菌的高效多基因改造。因此,本发明还请求保护所述方法在构建多基因改造酵母菌方面的应用。
31、具体地,所述酵母菌为熊蜂生假丝酵母菌。
32、作为一种实施方案,本发明还提供了一种多基因改造的酵母菌,所述改造的酵母菌是以熊蜂生假丝酵母菌为出发菌株,按照本发明所述方法进行多基因改造而得。
33、具体地,所述多基因改造是敲除酵母菌的mfo-2基因并过表达mrd-2和pugtb基因。
34、具体地,所述mfo-2基因的核苷酸序列如seq id no.1所示;mrd-2基因的核苷酸序列如seq id no.5所示;pugtb基因的核苷酸序列如seq id no.6所示。
35、作为一种可选地实施方案,本发明敲除mfo-2基因所用mfo-2基因的上游同源臂的核苷酸序列如seq id no.2所示,mfo-2基因的下游同源臂的核苷酸序列如seq id no.3所示。
36、具体地,所述敲除了mfo-2基因并过表达了mrd-2和pugtb基因的多基因改造熊蜂生假丝酵母菌的构建方法包括以下步骤:
37、s1.通过pcr扩增得到带有同源序列的mfo-2基因的上下游同源臂的dna片段,以及mrd-2和pugtb基因的dna片段;
38、s2.以s1所得dna片段为模板,通过两步法融合pcr和巢氏pcr将所有dna片段连接成用于多基因改造的长双链dna目标载体;
39、s3.将s2所得长双链dna目标载体单链化为长单链dna目标载体并转化酵母菌感受态细胞,筛选阳性转化子。
40、具体地,本发明所述用于扩增mfo-2基因的上游同源臂的引物为mfo-2_5f和mfo-2_5r,其序列依次如seq id no.7~8所示;用于扩增mfo-2基因的下游同源臂的引物为mfo-2_3f和mfo-2_3r,其序列依次如seq id no.9~10所示;用于扩增mrd-2基因的引物为mrd-2oef和mrd-2oer,其序列依次如seq id no.11~12所示;用于扩增pugtb基因的引物为pugtb oef和pugtb oer,其序列依次如seq id no.13~14所示。
41、具体地,扩增mfo-2基因的上游同源臂的pcr反应体系为:primestar max premix25.0μl,基因组dna2.0μl,上下游引物各1.0μl,dd h2o补足50μl;反应程序为:98℃10sec;55℃5sec,72℃5sec/kb计算延伸时间,30个循环;72℃10min。
42、扩增mfo-2基因的下游同源臂、mrd-2基因和pugtb基因的pcr反应体系与扩增mfo-2基因的上游同源臂相同,反应程序也相同。
43、具体地,扩增各dna片段时,基因组dna的用量为50~200ng。
44、具体地,本发明在构建用于改造酵母菌的长双链dna目标载体时还引入了改造后的用于抗性筛选的抗性基因,其核苷酸序列如seq id no.4所示,为潮霉素抗性基因。
45、在本发明中,所述改造后的抗性基因的dna片段是通过合成得到。
46、具体地,上述基因片段进行融合pcr时的连接顺序为:mfo-2基因的上游同源臂+pugtb+抗性筛选基因+mrd-2+mfo-2基因的下游同源臂;
47、具体地,上述基因片段的两步法pcr融合过程为:将带有同源臂的各基因片段以相同摩尔量加入pcr反应体系,总dna量为500~1000ng,不添加任何引物,进行5~10个循环,延伸速度为30~60s/kb,完成第一步融合pcr。
48、优选地,总dna量为800ng,10个循环,延伸速度为30s/kb。
49、向第一步融合pcr的pcr产物中加入引物mfo-2_5f和mfo-2_3r,进行25~30个循环,延伸速度为30~60s/kb,完成第二步融合pcr。
50、具体地,所述多基因改造酵母菌进行巢氏pcr所用引物的序列依次如seq idno.15~16所示;seq id no.16所示引物的5’端修饰有磷酸基团。
51、具体地,所述多基因改造酵母菌进行巢氏pcr的过程为:取第二步融合pcr的产物5~10μl稀释100~500倍,取稀释后的产物2~10μl为模板,加入seq id no.15~16所示引物,进行25~30个循环的巢式pcr,延伸速度为5~10s/kb,扩增得到反义链5’端是磷酸基团的长双链dna线性目标载体。
52、优选地,取第二步融合pcr的产物10μl,稀释200倍,取5μl作为巢式pcr模板,延伸速度为5s/kb。
53、本发明具有以下有益效果:
54、本发明提供了一种高效构建多基因改造酵母菌的方法,该方法通过两步法融合pcr及巢氏pcr构建得到长度达8000bp以上的长双链dna目标载体,并将其单链化为长单链dna目标载体,从而高效成功转化酵母菌,大大缩短了构建载体所需时间,提高了长链dna转化酵母菌体的成功率,提高了酵母菌的改造效率,步骤简单易操作、耗时短、载体转化效率高,成本低,可广泛用于酵母菌的多基因改造。本发明利用该方法构建了敲除mfo-2基因并过表达mrd-2和pugtb基因的多基因改造熊蜂生假丝酵母菌,其与野生型相比可在更短时间内发酵生成等量的槐糖脂,有利于在实际生产中缩短发酵用时,节省能耗,提高槐糖脂的发酵效率。
- 还没有人留言评论。精彩留言会获得点赞!