一种舒巴坦酸的制备方法与流程
本发明涉及药物合成,特别是涉及一种舒巴坦酸的制备方法。
背景技术:
1、舒巴坦酸,化学名(2s-cis)-3,3-二甲基-7-氧代-4-硫代-1-氮杂双环[3,2,0]庚烷-2-羧酸-4,4-二氧化物,分子式为c8h11no5s,分子量为233.24。
2、结构式:
3、
4、舒巴坦酸是一种不可逆的竞争性β-内酰胺酶抑制剂,本身抗菌活性弱,但与青霉素类或头孢菌素类药物联用,则有明显的协同作用,不仅提高了抗菌活性,也扩大了抗菌谱。目前国内生产舒巴坦酸的方法主要有两种:方法一、加压氢化反应,如cn102503957a中公开了在6,6-二溴青霉烷砜酸(dbpas)还原脱溴制备舒巴坦反应过程中,以雷尼镍为催化剂,在0.5~0.8mpa氢气的压力下,以液相介质循环射流的方式,对二溴青霉烷砜酸进行脱溴还原制备出舒巴坦酸,其不足之处在于:用氢气加压进行还原脱溴,安全风险较大;方法二、金属还原剂在有机溶剂和水的混合液中进行还原反应,如cn116143802a中公开了一种6,6-二溴青霉烷砜酸(dbpas)作为起始物料,乙酸乙酯、纯化水作为溶剂,使用锌粉还原制备舒巴坦酸的方法,其不足之处在于:6,6-二溴青霉烷砜酸(dbpas)和锌粉在有机溶剂中,6,6-二溴青霉烷砜酸(dbpas)形成自由基,发生聚合反应后,生成大分子有色杂质,影响产品的收率、质量和稳定性,有机溶剂中产出的舒巴坦酸外观不白,且随时间变化,颜色加深明显。
5、因此,研制一种更安全、环保、产物收率高、纯度高、稳定性好、外观好的适合工业化的制备舒巴坦酸的方法是目前亟待解决的新课题。
技术实现思路
1、为了克服上述现有技术的不足,本发明提供了一种水环境制备舒巴坦酸的合成方法,避免了自由基聚合等副反应的发生,大大提高了产品的纯度、收率和稳定性,该方法为常压反应,提高制备过程的安全性。
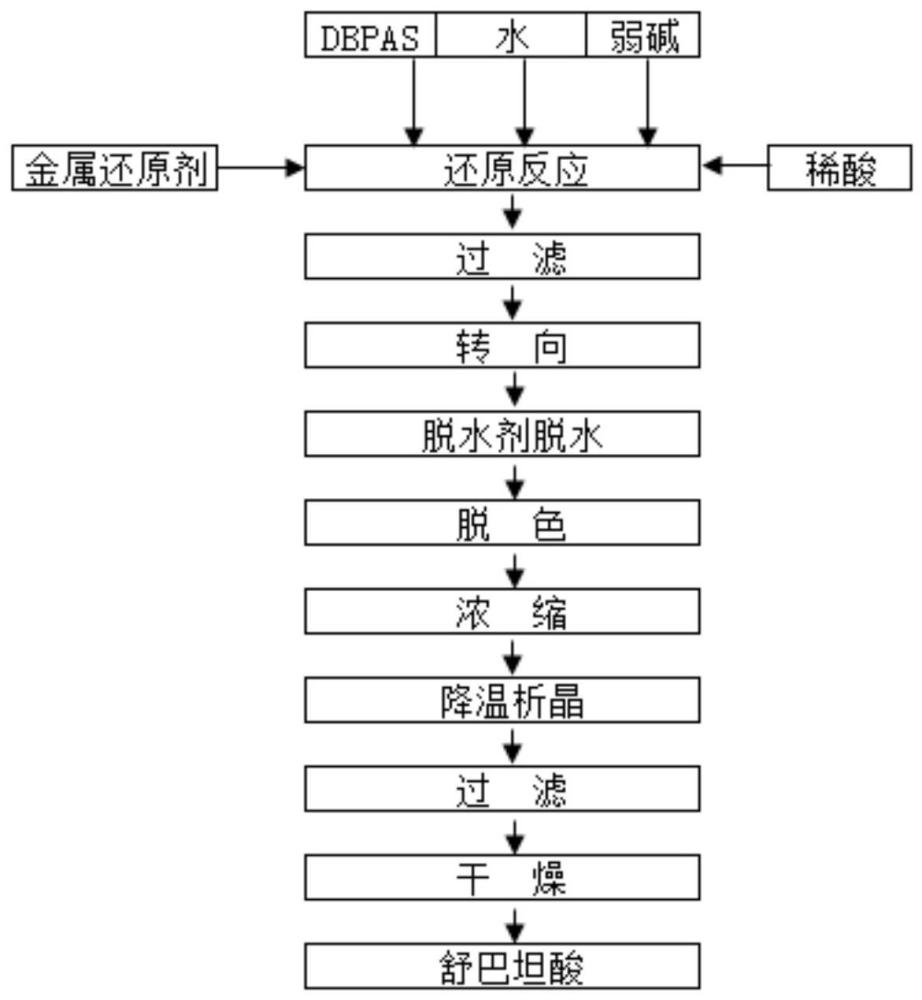
2、本发明所采用的技术方案是:一种舒巴坦酸的制备方法,所述制备方法包括以下步骤:
3、(1)6,6-二溴青霉烷砜酸和弱碱在无机溶剂中进行成盐反应,随后缓慢加入还原剂和第一稀酸水溶液,控制体系ph,进行还原反应,过滤,得到还原反应液;所述无机溶剂选自水;
4、(2)所述还原反应液和第一有机溶剂混合后,向其中缓慢加入第二稀酸水溶液,调节溶液ph值,反应一段时间,过滤,滤液分层;
5、(3)取水层加入第二有机溶剂进行萃取,将第二有机溶剂层用脱水剂进行脱水,第一过滤,浓缩、析晶、第二过滤、干燥。
6、在所述步骤(1)中,所述弱碱选自碳酸氢钠、碳酸氢钾、碳酸钠、碳酸钾中的一种或几种;所述成盐反应的温度为0~15℃;所述成盐反应过程中的ph值为2.5-4.5;所述还原剂选自锌、镍中的一种或几种;所述第一稀酸选自稀盐酸、稀硫酸中的一种或几种;所述第一稀酸的浓度为4-12%;所述控制体系ph为将ph控制在3-5;所述还原剂和稀酸的加入时间为0.1-3小时;所述还原剂加入完毕后继续进行还原反应的时间为15-50分钟。
7、在所述步骤(2)中,所述第一有机溶剂选自乙酸乙酯、氯仿、乙酸甲酯、二氯甲烷中的一种或几种;所述第二稀酸选自稀盐酸、稀硫酸中的一种或几种;所述稀硫酸的浓度为25-30%;所述调节溶液ph值为调节ph值在0.6-1.5;所述反应一段时间为反应5-20分钟;所述反应的温度0~20℃。
8、在所述步骤(3)中,所述第二有机溶剂选自乙酸乙酯、氯仿、乙酸甲酯、二氯甲烷中的一种或几种;所述脱水剂选自溶液脱水剂、固体脱水剂中的一种;所述溶液脱水剂选自饱和氯化钙水溶液、饱和氯化钠水溶液中的一种;所述固体脱水剂选自无水硫酸钠;所述6,6-二溴青霉烷砜酸的质量与第二有机溶剂的体积比为30:100-300,优选的,所述6,6-二溴青霉烷砜酸的质量与第二有机溶剂的体积比为30:150-250,所述质量的单位为g,所述体积的单位为ml;所述6,6-二溴青霉烷砜酸的质量与液体脱水剂的体积比为30:30-90,优选的,所述6,6-二溴青霉烷砜酸的质量与液体脱水剂的体积比为30:45-75;在所述第二过滤后,干燥前还包括采用所述第二有机溶剂洗涤物料的步骤。
9、在所述步骤(1)中,所述6,6-二溴青霉烷砜酸的质量与无机溶剂的体积比为30:150-300,优选的,所述6,6-二溴青霉烷砜酸的质量与无机溶剂的体积比为30:175-250,所述质量的单位为g,所述体积的单位为ml;所述6,6-二溴青霉烷砜酸与还原剂的质量比为30:11-12.5,优选的,所述6,6-二溴青霉烷砜酸与还原剂的质量比为30:11-12;所述6,6-二溴青霉烷砜酸的质量与第一有机溶剂的体积比为30:125-200,优选的,所述6,6-二溴青霉烷砜酸的质量与第一有机溶剂的体积比为30:140-170,所述质量的单位为g,所述体积的单位为ml。
10、在所述步骤(2)中,所述反应一段时间之后,过滤之前,还包括第一活性炭脱色的步骤;所述第一活性炭脱色的时间为0.5-2小时,优选的,所述第一活性炭脱色的时间为0.7-1.5小时;所述第一活性炭脱色的温度为15~30℃,优选的,所述第一活性炭脱色的温度为18~25℃;所述6,6-二溴青霉烷砜酸与第一活性炭的质量比为30:0.1-2,优选的,所述6,6-二溴青霉烷砜酸与第一活性炭的质量比为30:0.2-1。
11、在所述步骤(3)中,在所述将第二有机溶剂层用脱水剂进行脱水后,第一过滤前,还包括第二活性炭脱色的步骤;所述第二活性炭脱色的时间为0.5-2小时,优选的,所述第二活性炭脱色的时间为0.7-1.5小时;所述第二活性炭脱色的温度为-2~6℃,优选的,所述第二活性炭脱色的温度为-1~5℃;所述6,6-二溴青霉烷砜酸与第二活性炭的质量比为30:0.1-2,优选的,所述6,6-二溴青霉烷砜酸与第二活性炭的质量比为30:0.2-1;所述浓缩选自减压浓缩;所述减压浓缩的温度为20~35℃,优选的,所述减压浓缩的温度为25~30℃;所述减压浓缩的真空度为-0.09~-0.1mpa;所述浓缩得到浓缩液,所述6,6-二溴青霉烷砜酸的质量与浓缩液的体积比为30:30-60,优选的,所述6,6-二溴青霉烷砜酸的质量与浓缩液的体积比为30:35-50,所述质量的单位为g,所述体积的单位为ml;所述析晶的温度为-10~10℃,优选的,所述析晶的温度为-5~5℃;所述析晶的时间为10-30分钟,优选的,所述析晶的时间为15-25分钟;所述干燥选自真空干燥;所述真空干燥的温度选自10~40℃,优选的,所述真空干燥的温度选自25~35℃;所述真空干燥的时间为0.5-2小时,优选的,所述真空干燥的时间为0.8-1.2小时;所述真空干燥的真空度为-0.09~-0.1mpa。
12、在所述步骤(1)中,所述6,6-二溴青霉烷砜酸和弱碱在无机溶剂中进行成盐反应的具体步骤包括所述无机溶剂和6,6-二溴青霉烷砜酸混合后加入所述弱碱调节ph;所述无机溶剂和6,6-二溴青霉烷砜酸混合后的混合液的温度为0~10℃。
13、在所述步骤(1)中,所述成盐反应的温度为0~10℃;所述成盐反应过程中的ph值为3-4;所述第一稀酸的浓度为5-10%;所述控制体系ph为将ph控制在3.5-4.5;所述还原剂和稀酸的加入时间为0.5-2小时;所述还原剂加入完毕后继续进行还原反应的时间为20-40分钟。
14、在所述步骤(2)中,所述反应一段时间为反应5-15分钟;所述反应的温度5~15℃。
15、本发明所采用的技术方案的原理是:(1)采用水环境制备合成舒巴坦酸,避免了自由基聚合等副反应的发生,大大提高了产品的纯度、收率和稳定性,降低了杂质含量,改善了产品的吸光度和外观;(2)通过控制成盐反应过程中的ph值为3-4左右,可辅助改善产品的纯度、收率、稳定性、杂质含量、产品的吸光度等;(3)通过对成盐反应过程中无机溶剂水的用量进行控制,可辅助改善产品的外观,且在本技术限定的可选范围内,水的用量越大,产品外观越好;(4)在还原反应步骤中,在本技术限定的可选浓度范围内,所用的第一稀酸的浓度较低时,产品外观更好、收率更高、杂质含量越小;(5)通过在还原反应过程中控制ph以及控制还原剂的加入时间,可辅助改善反应收率和产品外观等;(6)通过在本技术限定的脱色温度范围内进行第一活性炭脱色的步骤,以及在本技术限定的脱色温度范围内进行第二活性炭脱色的步骤,可辅助改善产品的外观,尤其是第一活性炭脱色步骤对于改善产品外观的作用更大。
16、与现有技术相比,本发明的有益效果是:
17、1.本发明的目的在于避免现有技术的不足,在源头控制杂质的产生,提高产品质量、收率、稳定性,改善产品外观,为下游产品提供良好的质量基础。
18、2.本发明所述方法得到的舒巴坦酸,提升了产品质量,降低了杂质含量,得到的产品外观洁白晶莹,室温放置产品不再发黄,优选条件下,吸光度可达0.002,使用舒巴坦酸含量杂质ep方法检测,纯度可达99.8%,最大非特定杂质0.01%,其他杂质总和0.05%。
19、3.本发明所述方法得到的舒巴坦酸,稳定性好,在室温条件下,放置6个月,产品质量无明显变化;优选条件下,其产品含量仍保持99.5%以上,产品颜色无明显变化,保持白色。
20、4.本发明所述方法大大提高了舒巴坦酸的收率,优选条件下的收率高达91%以上,较有机反应环境提高10%。
21、5.本发明提高了舒巴坦酸生产过程的安全性。
- 还没有人留言评论。精彩留言会获得点赞!