一种用于百草枯检测的碳量子点荧光探针的制备方法与流程
本发明涉及材料科学领域,特别涉及具有对百草枯检测的碳量子点荧光探针的制备方法。
背景技术
百草枯,化学名称是1-1-二甲基-4-4-联吡啶阳离子盐,是一种快速灭生性除草剂,具有触杀作用和一定内吸作用。能迅速被植物绿色组织吸收,使其枯死。百草枯对人毒性极大,成人致死量为20%水溶液5~15毫升(20~40mg/kg),且无特效解毒药,口服中毒死亡率可达90%以上。目前已被20多个国家禁止或者严格限制使用。随着除草剂的广泛应用,百草枯残留中毒现象日渐普遍,并已成为继急性有机磷中毒之后的常见农药中毒,且中毒人数呈逐年上升的趋势。由于百草枯的致死用量极低与其无特效解药的特性,因此,百草枯残留的检测对方法的灵敏性和准确性要求越来越高。
目前已有的测定方法可概括为三类:活生物体分析法、仪器分析法、免疫化学分析法(食品工业科技,2012,33(18):393-396)。其中活生物体分析法利用自然界筛选对生长环境中有毒农药敏感的物种,根据其在含有有毒化合物的水体中的生长率变化来检测目标农药浓度大小(bull.environ.contam.toxicol.,2001,67:233-238)。但这种方法耗时较长,结果重复性较低,不能很好适用于如今百草枯残留检测需要。
仪器分析法包含气相色谱法(gc)、高效液相色谱法(hplc)、质谱联用法、毛细管电泳法(ce)、分光光度计法等(农药,2014,53(1):4-6),其中气相色谱法具有分离效果高、检测灵敏度高、选择性好等优点,是分析检测中比较常用的方法。由于gc法只能检测挥发性物质,而百草枯因为极性强、难以气化,所以必须通过化学衍生的方法转化为具挥发性物质才能进行分析。已经报道的衍生法中,硼氢化钠还原法较易推广应用,也是目前gc法中主要采用的处理方法。张婷等通过在碱性条件下,用硼氢化钠将百草枯还原成三级胺进行全血中的百草枯gc法检测,其线性相关系数为0.9965,检测限达20μg/l,回收率为99.3%±7%(广东公安科技,2007,89(4):21-22)。khan用硫酸对样品进行提取处理后,再经氧化铝柱钝化以对莴苣、萝卜、洋葱中百草枯进行gc检测,最小检测限达0.005mg/kg,标准偏差为4%(bull.environ.contam.toxicol.,1975,14(6):745-749)。通常情况下百草枯较难气化裂解,所以用气相色谱法检测百草枯操作较复杂,需要训练有素的操作人员,不适合检测大批量样品。
液相色谱法及液-质联用法液相色谱法具有快速、灵敏、准确的特点,也是目前技术相对成熟的一种检测手段。液相色谱适用于检测热稳定性差,极性强、分子量大、不易挥发的化合物及离子型化合物。百草枯是一种极性很强的离子型化合物,比较适合用此法进行分析。王朝虹等建立了lc/ms/ms检测生物体液中百草枯的方法,应用离子交换固相萃取法提取百草枯并检测,此法测得百草枯的最小检出限为10ng/ml血(s/n≥3),线性范围为0.02~20μg/ml。该方法快速、灵敏、准确,适用于生物检材中百草枯的分析(中国法医学杂志,2008,23(2):114-115)。yuangaozou等以二乙基百草枯为内标物,在紫外检测器波长256nm下检测,准确率为97.6%~107.3%,回收率为91.9%,相关性系数为0.9984,检出限为0.02~10μg/ml,此法简单灵敏高效(j.chromatographyb,2001,879:1809-1812)。刘育清等用aq-c18色谱柱和pda检测器,以kh2po4缓冲溶液(ph=1.9)为流动相,在290nm波长下,对百草枯的二氯盐进行定量分析。其结果标准偏差、变异系数为0.069和0.23%,相关系数为0.9998,回收率在98.91%~100.87%,此法分离效果好,准确度与精密度高,线性范围较宽,操作简便(第九届全国农药质量管理与分析技术交流会-论文集:170-173)。秦叶民等使用alltimac18色谱柱,流动相为乙腈/0.02mmol/l己烷磺酸钠溶液(体积比为65∶35),紫外检测器波长为254nm,测定结果的相对标准偏差为rsd=0.13%,回收率为98.9%~99.5%(第9届全国离子色谱学术报告会论文集,2002)。王瑞花等以十二烷基三甲基溴化铵和十二烷基硫酸钠预处理过的c18固相柱萃取,hplc/dad进行分析。结果回收率为81%~94%,检出限为1ng/ml,线性范围为50ng/ml~1mg/ml(法医学杂志,2005,21(2):121-123)。从上可以看出,液相色谱法是目前主流的检测百草枯的方法,它的优点很突出,但是同时也应注意它不适合处理大量样品。毛细管电泳法是一种高效便捷、快速、低消耗的新型检测技术。目前,ce应用与百草枯检测的报道还比较少。成美容等建立了毛细管电泳-间接化学发光法对茶叶中残留的百草枯进行检测,结果显示,在百草枯浓度为5×10-7~5×10-5mol/l范围内线性良好,线性相关系数为0.9945,最低检出限为9.4×10-8mol/l(分析测试学报,2009,28(12):1444-1447)。unez等用毛细管法检测水中的百草枯,以多孔石墨固相柱萃取为检材,用压力方式进样,用二极管阵列检测器进行检测,百草枯的回收率只有约40%(j.chromatographya,2002,946(1/2):275-282),结果不理想,有待于进一步研究。虽然毛细管电泳法具有样品前处理简单、避免分析柱损伤等优点,但准确性较差,检测限较高。
免疫化学分析法包括放射免疫法(ria)、酶联免疫法(elisa)和胶体金免疫检测(gica)等(农药,2014,53(1):4-6),虽然这些技术具有灵敏度高、稳定性好、特异性强等特点,但这些方法需要的试剂、设备价格昂贵,且具有一定的危险性,其应用受到很大限制。而且在实际操作中很容易受操作条件影响,易出现假信号,从而影响了结果的准确性。
上述方法优点甚多,但其制备步骤繁琐,成本高,且某些检测方法对大型仪器的依赖性强,无法满足便捷和现场检测的需求。
近年来,对痕量百草枯检测技术的越来越多,2011年王勇等人公开了发明专利(cn201110186707.2)“一种测定百草枯血药浓度的方法”。该发明采取以下步骤:1、取血浆样品,加入内标物的水溶液和蛋白沉淀剂乙腈,旋涡混合,再离心后取上清液进样;2、采用通用型c18液相色谱柱,高效液相系统采用通用型的二极管阵列检测器及高压泵,酸性流动相使用离子对试剂,所述的酸性流动相为3mmol•l-1十二烷基磺酸钠水溶液-0.2%三氟乙酸水溶液-乙腈-水的混合液,等梯度洗脱;3、采用二极管阵列检测器进行检测,检测波长为250~260nm,测定内标物和百草枯的峰面积,以最小二乘法线形回归计算出百草枯的血药浓度。2011年,杜晶晶等公开了发明专利(cn200910241254.1)“一种新型的百草枯检测方法”。该发明采用以下步骤:1、利用硅烷修饰fe3o4表面后,将其分散于硝酸银溶液中,在还原剂盐酸羟胺的作用下,制备得到核壳式fe3o4/ag磁性纳米颗粒,这种复合颗粒兼具fe3o4的磁性与纳米银颗粒的拉曼增强性能。2、将制备出的fe3o4/ag磁性纳米颗粒分散到一定浓度的百草枯溶液中,由于百草枯分子的主体官能团与磁性纳米颗粒表面存在化学作用,因此它可以迅速被吸附在磁性颗粒外层银壳表面,利用外加磁场将分散在溶液中的磁性颗粒进行回收后,通过便携式拉曼光谱仪即可以检测到百草枯的拉曼特征峰。2013年关杰等人公开了发明专利(cn201310530983.5)“一种环境中百草枯残留量的检测方法”。该发明采取以下步骤:1、将待测土壤样品除去固体杂质、干燥、粉碎;2、将步骤1所得样品加入酸和乙醇浸泡6~12h,过滤,等体积萃取三次,浓缩萃取相;3、将步骤2所得样品用正相柱纯化,将含百草枯的馏分蒸干;4、将步骤3所得样品用反相柱检测,用百草枯标准曲线计算百草枯浓度。2014年刘靖靖等人公开了发明专利(cn201410096085.8)“一种食品中百草枯和敌草快残留量的测定方法”。该发明采取以下步骤:1、称取均质化的食品样品于具塞塑料离心管中,加甲醇水溶液涡旋振荡,离心;2、获得的上清液于塑料离心管中,加入基质分散固相萃取剂,涡旋振荡,离心,吸取上清液过0.22μm滤膜,所得样品液待测定;3、将空白样品按上述步骤(1)、(2)处理,用该空白样品基质溶液在10~1000μg/l范围内配制百草枯和敌草快系列浓度标准工作液;4、液相色谱--四极杆飞行时间串联质谱(lc-qtof)测定。2015年梁秦公开了发明专利(cn201510464217.2)“一种快速定量检测病人血液中百草枯浓度的方法”。该发明采用以下步骤:1、标准曲线绘制配制500μg/ml的百草枯标准溶液,将其用健康人血清逐级稀释为10、5、2.5、1、0.2、0.08、0.005μg/ml的百草枯溶液,与乙腈和20%三氯乙酸溶液按照三者体积比6∶1∶1.3混匀,以12000r/min离心10min,取100μl上清液,加1900μl蒸馏水稀释,12000r/min离心10min取上清,以蒸馏水为空白对照,采用紫外分光光度法进行测定,在257nm测定吸光度值(y)并对质量浓度(x)进行回归,得线性回归方程y=0.0935x+0.2964,其相关系数为0.9955。2、取离心好的样品血清600μl,先加入100μl除蛋白试剂溶剂i,再加入130μl配好的试剂ⅱ,轻轻混匀;12000r/min离心10min;取上清液,以蒸馏水调零,在257nm波长下用紫外分光光度仪(或全自动/半自动生化分析仪、酶标仪等)测定吸光度,代入上述方程y=0.0935x+0.2964,得样品血清中百草枯浓度(μg/ml)。
虽然上述发明有较多可取之处,但是这些方法都存在试样处理较为繁琐,成本高,选择性差,灵敏度低。而荧光探针的方法可以克服以上缺陷。在近年的实用检测技术中,荧光探针已经越来越受到科研人员的关注,广泛应用于药物控释、靶向给药、检测药效和各种含有特定基团的大分子的检测。所以,在荧光探针的材料选择上,溶解性较好,在光的照射下很难分解、发光范围广、强度大、价格便宜、易于制备的碳量子点逐渐成为广大研究者青睐的材料,得到了广泛的关注。
霍丹群等人公开了发明专利(cn201510449999.2)“一种用于检测氟啶胺的试纸及检测方法”公开一种用于检测氟啶胺的试纸及检测方法,以半胱氨酸为碳源,采用水热法制备碳量子点溶液,再将不含荧光剂的滤纸置于碳量子点溶液中浸泡并干燥,制得试纸;检测方法为先在上述试纸上点样不同含量的氟啶胺和空白试剂,并在晾干后于紫外光下观察并拍照,制成标准比色卡,再将样品点样于试纸上,晾干后在紫外光下与比色卡进行颜色深浅的对比,半定量出样品中的氟啶胺含量;具有分析成本低、操作简便快速、选择性高、稳定性好、实现了可视化检测的优点。
杨秀培等人公开了发明专利(cn201510456220.x)“一种利用芦荟为碳源制备碳量子点的及柠檬黄检测方法”。利用新鲜芦荟和超纯水,180℃反应11小时,自然冷却至室温后过滤,向滤液中加入等体积二氯甲烷除去未反应有机物,收集上层浅黄色水溶液,装入透析袋中纯化后,得到碳量子点水溶液,置于4℃保存备用。所得碳量子点荧光量子产率高且具有强而稳定的荧光性质。利用柠檬黄分子对碳量子点选择性高效的荧光淬灭效应,该量子点在柠檬黄的检测中所用仪器便宜、试剂耗费少、耗时少、方法准确度和重现性均很好。
高大明等人公开了发明专利(cn201410615249.3)“对痕量百草枯检测的cdte量子点荧光探针的化学制备方法”,其特征在于:所述的cdte量子点表面带有羧基,其表面带负电荷的羧基与带正电荷的目标分子百草枯通过正负电荷的静电作用,当cdte量子点与目标分子百草枯在空间上相互接近,通过荧光共振能量转移原理,cdte量子点荧光探针发射光谱为红色发光谱带能够被绿色的目标分析物百草枯分子所吸收,利用cdte量子点荧光强度的改变,实现对痕量百草枯的检测,本发明的制备过程包括如下两个步骤:首先制备紫色透明的nahte溶液,其次在发射光谱带为红色的cdte量子点荧光探针的表面修饰巯基乙酸后调节ph在10~12之间,氮气环境下加入制备的nahte溶液,并控制回流,得到不同荧光发光谱带的表面修饰了羧基的cdte量子点。最后将得到的产物用丙酮清洗三次去除多余未反应底物后重新分散在去离子水中,得到表面带负电荷羧基的cdte量子点荧光探针,这种红色发光谱带的cdte量子点荧光探针对百草枯具有选择性、灵敏性,实现对百草枯的痕量探测。其中表面修饰了羧基的cdte量子点荧光探针,能够对百草枯分子选择性识别。当加入一定量的百草枯目标分子后,发射光谱带为红色的cdte量子点富电子羧基能够与缺电子百草枯通过正负电荷静电作用,绿色百草枯分子正好吸收cdte量子点的所发射红色光谱带,从而导致荧光强度下降,实现对百草枯的检测。其次与传统的农药检测方法相比,表面修饰羧基的cdte量子点的荧光探针具有较大的比表面积,拥有更多的识别位点,提高对目标分子选择性识别,利用荧光共振能量转移原理,提高了对目标分析物的高敏感的痕量检测。并且cdte量子点粒径和厚度可控,可以调节回流反应时间来加以控制。此过程制备出的cdte量子点荧光探针过程较为繁琐,cdte量子点本身存在cd2+离子,容易在量子点表面解离释放出来产生毒性,在氧化性环境和紫外线照射时这种效应更加明显,以及外界条件和量子点本身的一些性质都会影响到量子点的稳定性,荧光容易漂白,易对环境造成二次污染。
可见上述方法中制备的量子点荧光探针都存在缺陷和不足,远不如碳量子点荧光探针制备简便。同时,对于碳量子点荧光探针对百草枯的检测未见报道,因此,合成高选择性和高灵敏的表面富含羟基和羧基的碳量子点荧光探针,简单便捷地实现对痕量百草枯分子识别和检测有其必要性。
在本发明中,我们报道了基于荧光共振能量转移原理合成了一种用于痕量百草枯检测表面富含羟基和羰基的碳量子点荧光探针的制备方法。所制备的碳量子点荧光探针表面带有负电荷的羟基和羧基可直接通过阴阳离子静电的相互作用与带正电荷的百草枯进行特异性结合,在空间相互接近时,发红色光谱带的碳量子点荧光探针发射光谱能够被自然光下绿色的目标分子所吸收,发生荧光共振能量转移,导致碳量子点荧光探针荧光强度降低,实现对目标分子选择性识别和检测。
因此,本发明所制备的用于痕量百草枯检测的碳量子点荧光探针,其具有制备步骤简单,选择性强,灵敏性高,结合位点多,结合容量大,结合动力学速度快,可重复使用,便捷低廉等优点。
技术实现要素:
发明目的:针对目前现有技术存在的不足之处,本发明利用间苯二胺作为前驱体制备出碳量子点荧光探针。该碳量子点荧光探针中碳量子点粒径大部分分布在2~3nm范围内,表面带负电荷。由于表面带负电荷的羟基和羧基的碳量子点荧光探针与带正电荷的百草枯分子通过阴阳离子对静电的相互作用,进行特异性结合,在两种空间互相接近时,发生荧光共振能量转移,导致碳量子点荧光探针荧光强度改变,从而实现对目标分子百草枯选择性识别和检测。所述合成方法为溶剂热法。本发明以间苯二胺溶解在无水乙醇中,在干燥箱内程序升温至恒温反应后、自然冷却至室温,经层析色谱分离后,透析袋透析,制备出对百草枯具有高选择性、高灵敏性识别和痕量检测作用的碳量子点荧光探针。
本发明的技术方案是:一种用于百草枯检测的碳量子点荧光探针的制备方法,其特征在于:所述的碳量子点荧光探针发射光谱带为红色荧光,其表面含有羟基和羧基的亲水的基团,量子点荧光探针表面带负电的羟基和羧基与带正电目标分子在空间相互接近时,通过阴阳离子对静电的相互作用,发红色光谱带的碳量子点荧光探针发射光谱能够被自然光下绿色的目标分子所吸收,发生荧光共振能量转移,导致碳量子点荧光探针荧光强度降低,实现对目标分子选择性识别和检测,所述的碳量子点荧光探针制备过程包括如下两个步骤:
第一步是碳量子点荧光探针的前驱液的制备:首先,准确的称取0.02~0.04g的前驱体溶解在30~50ml的无水乙醇中,溶解后,移入到体积为100ml的聚四氟乙烯内胆的反应釜中,用材质为聚四氟乙烯盖密闭,金属盖密封,将其置于程序升温速率为5℃/min的干燥箱中,升温至90~110℃的范围内反应1h后,再升温至150~170℃的范围内反应12h后,自然冷却至室温,取出反应釜,将反应产物倒入烧杯中,得到碳量子点荧光探针的前驱液;
第二步是碳量子点荧光探针的制备:首先,用层析色谱法进行分离,把填充好的硅胶柱竖直安装在铁架台上,向上述前驱液的烧杯里中加入40~60ml配好比例的展开剂,用胶头滴管吸取的前驱液,沿着柱壁以2ml/min速度滴加前驱液,结束后,再沿着柱壁加入以2ml/min的速度滴加20~40ml配好比例的淋洗剂,将层析分离后的产物再用透析袋透析,保留液用10~30ml去离子水稀释,得到表面含有羟基和羰基的碳量子点荧光探针。
作为对现有技术的进一步改进,本发明所述的目标分子是百草枯分子;所述的对目标分子选择性识别是通过阴阳离子对静电的相互作用;所述的对目标分子检测是基于荧光共振能量转移原理;所述的碳量子点荧光探针制备中的前驱体是间苯二胺;所述的碳量子点荧光探针制备中的展开剂是体积比为2:3的乙酸乙酯和石油醚混合液;所述的碳量子点荧光探针制备中的淋洗剂是体积比为1:5的乙酸乙酯和石油醚混合液;所述的碳量子点荧光探针制备方法为溶剂热法。
相对于现有技术的有益效果:
近年来,对痕量百草枯检测技术的越来越多,2011年王勇等人公开了发明专利(cn201110186707.2)“一种测定百草枯血药浓度的方法”。该发明采取以下步骤:1、取血浆样品,加入内标物的水溶液和蛋白沉淀剂乙腈,旋涡混合,再离心后取上清液进样;2、采用通用型c18液相色谱柱,高效液相系统采用通用型的二极管阵列检测器及高压泵,酸性流动相使用离子对试剂,所述的酸性流动相为3mmol•l-1十二烷基磺酸钠水溶液-0.2%三氟乙酸水溶液-乙腈-水的混合液,等梯度洗脱;3、采用二极管阵列检测器进行检测,检测波长为250~260nm,测定内标物和百草枯的峰面积,以最小二乘法线形回归计算出百草枯的血药浓度。2011年,杜晶晶等公开了发明专利(cn200910241254.1)“一种新型的百草枯检测方法”。该发明采用以下步骤:1、利用硅烷修饰fe3o4表面后,将其分散于硝酸银溶液中,在还原剂盐酸羟胺的作用下,制备得到核壳式fe3o4/ag磁性纳米颗粒,这种复合颗粒兼具fe3o4的磁性与纳米银颗粒的拉曼增强性能。2、将制备出的fe3o4/ag磁性纳米颗粒分散到一定浓度的百草枯溶液中,由于百草枯分子的主体官能团与磁性纳米颗粒表面存在化学作用,因此它可以迅速被吸附在磁性颗粒外层银壳表面,利用外加磁场将分散在溶液中的磁性颗粒进行回收后,通过便携式拉曼光谱仪即可以检测到百草枯的拉曼特征峰。2013年关杰等人公开了发明专利(cn201310530983.5)“一种环境中百草枯残留量的检测方法”。该发明采取以下步骤:1、将待测土壤样品除去固体杂质、干燥、粉碎;2、将步骤1所得样品加入酸和乙醇浸泡6~12h,过滤,等体积萃取三次,浓缩萃取相;3、将步骤2所得样品用正相柱纯化,将含百草枯的馏分蒸干;4、将步骤3所得样品用反相柱检测,用百草枯标准曲线计算百草枯浓度。2014年刘靖靖等人公开了发明专利(cn201410096085.8)“一种食品中百草枯和敌草快残留量的测定方法”。该发明采取以下步骤:1、称取均质化的食品样品于具塞塑料离心管中,加甲醇水溶液涡旋振荡,离心;2、获得的上清液于塑料离心管中,加入基质分散固相萃取剂,涡旋振荡,离心,吸取上清液过0.22μm滤膜,所得样品液待测定;3、将空白样品按上述步骤(1)、(2)处理,用该空白样品基质溶液在10~1000μg/l范围内配制百草枯和敌草快系列浓度标准工作液;4、液相色谱--四极杆飞行时间串联质谱(lc-qtof)测定。2015年梁秦公开了发明专利(cn201510464217.2)“一种快速定量检测病人血液中百草枯浓度的方法”。该发明采用以下步骤:1、标准曲线绘制配制500μg/ml的百草枯标准溶液,将其用健康人血清逐级稀释为10、5、2.5、1、0.2、0.08、0.005μg/ml的百草枯溶液,与乙腈和20%三氯乙酸溶液按照三者体积比6∶1∶1.3混匀,以12000r/min离心10min,取100μl上清液,加1900μl蒸馏水稀释,12000r/min离心10min取上清,以蒸馏水为空白对照,采用紫外分光光度法进行测定,在257nm测定吸光度值(y)并对质量浓度(x)进行回归,得线性回归方程y=0.0935x+0.2964,其相关系数为0.9955。2、取离心好的样品血清600μl,先加入100μl除蛋白试剂溶剂i,再加入130μl配好的试剂ⅱ,轻轻混匀;12000r/min离心10min;取上清液,以蒸馏水调零,在257nm波长下用紫外分光光度仪(或全自动/半自动生化分析仪、酶标仪等)测定吸光度,代入上述方程y=0.0935x+0.2964,得样品血清中百草枯浓度(μg/ml)。
虽然上述发明有较多可取之处,但是这些方法都存在试样处理较为繁琐,成本高,选择性差,灵敏度低。而荧光探针的方法可以克服以上缺陷。在近年的实用检测技术中,荧光探针已经越来越受到科研人员的关注,广泛应用于药物控释、靶向给药、检测药效和各种含有特定基团的大分子的检测。所以,在荧光探针的材料选择上,溶解性较好,在光的照射下很难分解、发光范围广、强度大、价格便宜、易于制备的碳量子点逐渐成为广大研究者青睐的材料,得到了广泛的关注。
霍丹群等人公开了发明专利(cn201510449999.2)“一种用于检测氟啶胺的试纸及检测方法”。以半胱氨酸为碳源,采用水热法制备碳量子点溶液,再将不含荧光剂的滤纸置于碳量子点溶液中浸泡并干燥,制得试纸;检测方法为先在上述试纸上点样不同含量的氟啶胺和空白试剂,并在晾干后于紫外光下观察并拍照,制成标准比色卡,再将样品点样于试纸上,晾干后在紫外光下与比色卡进行颜色深浅的对比,半定量出样品中的氟啶胺含量;具有分析成本低、操作简便快速、选择性高、稳定性好、实现了可视化检测的优点。
杨秀培等人公开了发明专利(cn201510456220.x)“公开了一种利用芦荟为碳源制备碳量子点的及柠檬黄检测方法”。利用新鲜芦荟和超纯水,180℃反应11小时,自然冷却至室温后过滤,向滤液中加入等体积二氯甲烷除去未反应有机物,收集上层浅黄色水溶液,装入透析袋中纯化后,得到碳量子点水溶液,置于4℃保存备用。所得碳量子点荧光量子产率高且具有强而稳定的荧光性质。利用柠檬黄分子对碳量子点选择性高效的荧光淬灭效应,该量子点在柠檬黄的检测中所用仪器便宜、试剂耗费少、耗时少、方法准确度和重现性均很好。
高大明等人公开了发明专利(cn201410615249.3)“对痕量百草枯检测的cdte量子点荧光探针的化学制备方法”,其特征在于:所述的cdte量子点表面带有羧基,其表面带负电荷的羧基与带正电荷的目标分子百草枯通过正负电荷的静电作用,当cdte量子点与目标分子百草枯在空间上相互接近,通过荧光共振能量转移原理,cdte量子点荧光探针发射光谱为红色发光谱带能够被绿色的目标分析物百草枯分子所吸收,利用cdte量子点荧光强度的改变,实现对痕量百草枯的检测,本发明的制备过程包括如下两个步骤:首先制备紫色透明的nahte溶液,其次在发射光谱带为红色的cdte量子点荧光探针的表面修饰巯基乙酸后调节ph在10~12之间,氮气环境下加入制备的nahte溶液,并控制回流,得到不同荧光发光谱带的表面修饰了羧基的cdte量子点。最后将得到的产物用丙酮清洗三次去除多余未反应底物后重新分散在去离子水中,得到表面带负电荷羧基的cdte量子点荧光探针,这种红色发光谱带的cdte量子点荧光探针对百草枯具有选择性、灵敏性,实现对百草枯的痕量探测。其中表面修饰了羧基的cdte量子点荧光探针,能够对百草枯分子选择性识别。当加入一定量的百草枯目标分子后,发射光谱带为红色的cdte量子点富电子羧基能够与缺电子百草枯通过正负电荷静电作用,绿色百草枯分子正好吸收cdte量子点的所发射红色光谱带,从而导致荧光强度下降,实现对百草枯的检测。其次与传统的农药检测方法相比,表面修饰羧基的cdte量子点的荧光探针具有较大的比表面积,拥有更多的识别位点,提高对目标分子选择性识别,利用荧光共振能量转移原理,提高了对目标分析物的高敏感的痕量检测。并且cdte量子点粒径和厚度可控,可以调节回流反应时间来加以控制。此过程制备出的cdte量子点荧光探针过程较为繁琐,cdte量子点本身存在cd2+离子,容易在量子点表面解离释放出来产生毒性,在氧化性环境和紫外线照射时这种效应更加明显,以及外界条件和量子点本身的一些性质都会影响到量子点的稳定性,荧光容易漂白,易对环境造成二次污染。
虽然上述发明有较多可取之处,但是这些方法都存在试样处理较为繁琐,成本高,专一性差,灵敏度低。而荧光探针的方法可以克服以上缺陷。在近年的实用检测技术中,荧光探针已经越来越受到科研人员的关注,广泛应用于药物控释、靶向给药、检测药效和各种含有特定基团的大分子的检测。所以,在荧光探针的材料选择上,溶解性解性较好,在光的照射下很难分解、发光范围广、强度大、价格便宜、易于制备的碳量子点逐渐成为广大研究者青睐的材料,得到了广泛的关注。而且这些方法制备出的检测材料合成步骤繁琐,成本高,专一性差,且某些样品检测预处理需要专业人士操作,不利于现场分析。而碳量子点荧光探针制备步骤简单,选择性高,灵敏性强,可重复使用且成本低廉,制备碳量子点荧光探针,实现对百草枯的高选择性识别和便捷检测未见文献给与报道,因此,合成高选择性、高灵敏的表面富含羟基和羧基的碳量子点荧光探针,实现对百草枯分子选择性的识别和敏感性的检测,有其重要的理论和实践意义。
本发明第一步是碳量子点荧光探针的前驱液的制备:首先,准确的称取0.02~0.04g的前驱体溶解在30~50ml的无水乙醇中,溶解后,移入到体积为100ml的聚四氟乙烯内胆的反应釜中,用材质为聚四氟乙烯盖密闭,金属盖密封,将其置于程序升温速率为5℃/min的干燥箱中,升温至90~110℃的范围内反应1h后,再升温至150~170℃的范围内反应12h后,自然冷却至室温,取出反应釜,将反应产物倒入烧杯中,得到碳量子点荧光探针的前驱液;
第二步是碳量子点荧光探针的制备:首先,用层析色谱法进行分离,把填充好的硅胶柱竖直安装在铁架台上,向上述前驱液的烧杯里中加入40~60ml配好比例的展开剂,用胶头滴管吸取的前驱液,沿着柱壁以2ml/min速度滴加前驱液,结束后,再沿着柱壁加入以2ml/min的速度滴加20~40ml配好比例的淋洗剂,将层析分离后的产物再用透析袋透析,保留液用10~30ml去离子水稀释,得到表面含有羟基和羰基的碳量子点荧光探针。
综上所述,表面富含羟基和羰基的碳量子点荧光探针,既增加碳量子点的荧光探针的比表面积,又增加了分子识别位点,还降低了毒性,对环境友好,提高了选择性、识别性和敏感性。
其二:表面富含羟基和羰基的碳量子点荧光探针,能够对百草枯分子选择性识别和检测。碳量子点荧光探针表面带负电荷的羟基和羧基基团与带正电荷的百草枯分子通过阴阳离子对静电的相互作用,形成了特殊性的识别,当目标分子百草枯与碳量子点荧光探针在空间上相互接近时,发红色光谱带的碳量子点荧光探针发射光谱能够被自然光下绿色的目标分子百草枯所吸收,发生荧光共振能量转移,导致碳量子点荧光探针荧光强度降低,实现对目标分子的检测。可见,本发明所提供的方法,实用范围比较广泛。
其三:与传统的有机荧光材料相比较,表面富含羟基和羰基的碳量子点荧光探针荧光寿命长,具有较大的比表面积,拥有较多的识别位点,提高对目标分子百草枯的选择性识别,利用荧光共振能量转移原理,提高了对目标分析物的高敏感的检测。
其四:选择表面富含羟基和羰基的碳量子点荧光探针的目的,因为其具有以下优点:(1)碳量子点荧光探针荧光寿命长;(2)溶剂热条件下合成相对简单,成本相对较低;(3)生物毒性低;(4)表面容易嫁接有机官能团;(5)对环境无害;(6)通过碳量子点荧光探针表面带负电荷的羟基和羧基基团与带正电荷的百草枯分子通过阴阳离子对静电的相互作用,形成了特殊性的识别,当目标分子百草枯与碳量子点荧光探针在空间上相互接近时,发红色光谱带的碳量子点荧光探针发射光谱能够被自然光下绿色的目标分子百草枯所吸收,发生荧光共振能量转移,导致碳量子点荧光探针荧光强度降低,实现对目标分子的检测。
附图说明
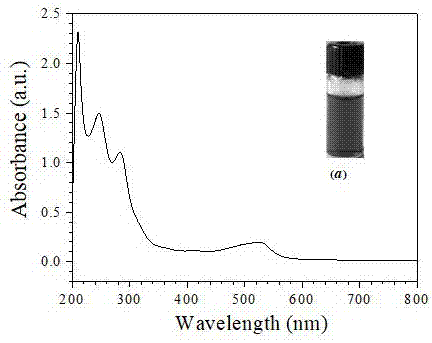
图1是本发明所采用的碳量子荧光探针紫外-可见吸收光谱。
图2是本发明所采用的归一化的碳量子点荧光探针的荧光光谱和百草枯的紫外-可见吸收光谱。
图3是本发明所采用的碳量子荧光探针红外光谱。
图4是本发明所采用的碳量子荧光探针粒径分布。
图5是本发明所采用的碳量子荧光探针的zeta电位。
图6是本发明所采用的碳量子荧光探针的拉曼光谱。
图7是本发明所采用的碳量子点荧光探针的荧光曲线随百草枯浓度改变的演变。
图8是本发明所采用的碳量子点荧光探针对不同浓度百草枯荧光光谱变化(a)及其所对应荧光淬灭常数(b)。
根据附图进一步解释具体实施方式
图1是本发明所采用的碳量子荧光探针紫外-可见吸收光谱。以间苯二胺溶解在无水乙醇中,溶解后转置聚四氟乙烯内胆的反应釜中,密封后,放置恒温干燥箱内,通过程序阶段升温,反应结束后、自然冷却至室温,经层析色谱分离后,透析袋透析,制备出对百草枯具有高选择性、高灵敏性识别和痕量检测作用的碳量子点荧光探针。图中插图1中插图(a)表示的是在自然光下碳量子点荧光探针。该碳量子点的紫外-可见特征吸收峰分别在250nm、295nm和520nm左右。
图2是本发明所采用的归一化的碳量子点荧光探针的荧光光谱和百草枯的紫外-可见吸收光谱。图2中(a)表示的是碳量子点荧光探针的归一化荧光发射光谱曲线,在630nm处荧光强度最大,(c)表示的在365nm紫外灯下碳量子点荧光探针发射光谱带为红色。图2中(b)表示的是百草枯水溶液的归一化紫外-可见吸收光谱曲线,在630nm处的吸收峰值最大,(d)表示的在自然光下百草枯水溶液呈现为绿色。给体(碳量子点)荧光发射光谱与受体(百草枯)可见吸收光谱的重叠程度是发生荧光共振能量转移的前提条件,图2中的归一化的碳量子点的荧光发射光谱(a)与百草枯的可见吸收光谱(b)完全重合,因此,碳量子点与百草枯之间可以发生荧光共振能量转移,导致碳量子点荧光探针发射荧光强度降低(或淬灭),实现碳量子点荧光探针对百草枯的检测。
图3是本发明所采用的碳量子荧光探针红外光谱。由图可知在3390cm-1处有-cooh的特征吸收峰;在2490cm-1处是c-h键的伸缩振动吸收峰;在1640cm-1处是-co-nh的伸缩振动吸收峰;在1770cm-1处是c=o的伸缩振动吸收峰;同时在1050-1360cm-1内有连续的c-o基团存在;在3400~3500cm-1处有-oh的特征吸收峰。结合以上分析,可以得到碳量子点表面存在大量的羧基和羰基。正是由于这些大量的亲水基团存在,恰恰说明了碳量子点的水溶性非常好。
图4是本发明所采用的碳量子荧光探针粒径分布。由图可知,碳量子点粒径呈现正态分布,碳量子点粒径大部分分布在2~3nm左右。
图5是本发明所采用的碳量子荧光探针的zeta电位。由图可知碳量子点表面电荷分布在-20~0mv范围内,大部分碳量子点表面电荷呈现-10mv左右。因此,表面带负电荷的碳量子点荧光探针能够与带正电荷的百草枯分子之间通过阴阳离子对静电的相互作用,实现对百草枯分子的特异性识别。
图6是本发明所采用的碳量子荧光探针的拉曼光谱。从图中可以看出在1345cm-1和1578cm-1处出现了较为明显的拉曼散射峰,分别对应着氧化石墨烯的d峰及g峰。d峰对应于物质结构的规整性,石墨被氧化后,结构中一部分sp2杂化碳原子转化成sp3杂化结构,即石墨层中的c=c双键被破坏;而g峰对应于e2g光学模的一阶拉曼散射,说明物质的结构非常规整。我们采用d/g的比值来衡量碳量子点荧光探针的规整度。比值越大,规整性越差,图中我们可以看出,d/g的比值较大,基本接近于90%,规整性很差,可以判定是碳量子点荧光探针,而不是石墨烯量子点荧光探针。
图7是本发明所采用的碳量子点荧光探针的荧光曲线随百草枯浓度改变的演变。分别取20μl的碳量子点荧光探针样品六份,再分别依次滴加浓度为0mol·l-1、1×10-9mol·l-1、1×10-8mol·l-1、1×10-7mol·l-1、1×10-6mol·l-1和1×10-5mol·l-1的百草枯水溶液,分别测定碳量子点荧光探针的荧光发射光谱,得到了碳量子点的荧光发射光谱自上而下变化曲线,可以看出随着百草枯浓度的不断增加,碳量子点荧光探针的荧光强度减弱,表明表面带负电荷碳量子点与带正电荷的目标分子百草枯之间通过阴阳离子对静电的相互作用,在彼此空间上相互接近时,发生荧光共振能量转移,从而使碳量子点荧光探针荧光强度下降,检测限达到1×10-9mol·l-1,成功实现了对百草枯选择性识别和痕量检测。
图8是本发明所采用的碳量子点荧光探针对不同浓度百草枯荧光光谱变化图(a)及其所对应荧光淬灭常数图(b)。图8中(a)中自上而下可以看到随着目标分子百草枯浓度自0mol·l-1、1×10-5mol·l-1、2×10-5mol·l-1、3×10-5mol·l-1、4×10-5mol·l-1、5×10-5mol·l-1、6×10-5mol·l-1、7×10-5mol·l-1增加至8×10-5mol·l-1,碳量子点的荧光强度逐渐减弱。根据stern-volme方程:(i0/i)-1=ksv·c,i0、i分别为没有目标分析物和存在目标分析物的稳态荧光强度,ksv为百草枯的淬灭常数,c表示百草枯水溶液浓度。图8中(b)的离散点拟合得到方程为:(i0/i)-1=0.07048·c,相对标准偏差为0.9940,相关系数为0.9970。因此,根据此拟合stern-volme方程可以得到百草枯的淬灭常数分别为:7048l·mol-1,淬灭常数数值反映了碳量子点荧光探针实现了对百草枯的检测。
具体实施方式:一种用于百草枯检测的碳量子点荧光探针的制备方法,其特征在于:所述的碳量子点荧光探针发射光谱带为红色荧光,其表面含有羟基和羧基的亲水的基团,量子点荧光探针表面带负电的羟基和羧基与带正电目标分子在空间相互接近时,通过阴阳离子对静电的相互作用,发红色光谱带的碳量子点荧光探针发射光谱能够被自然光下绿色的目标分子所吸收,发生荧光共振能量转移,导致碳量子点荧光探针荧光强度降低,实现对目标分子选择性识别和检测,所述的碳量子点荧光探针制备过程包括如下两个步骤:
第一步是碳量子点荧光探针的前驱液的制备:首先,准确的称取0.02~0.04g的前驱体溶解在30~50ml的无水乙醇中,溶解后,移入到体积为100ml的聚四氟乙烯内胆的反应釜中,用材质为聚四氟乙烯盖密闭,金属盖密封,将其置于程序升温速率为5℃/min的干燥箱中,升温至90~110℃的范围内反应1h后,再升温至150~170℃的范围内反应12h后,自然冷却至室温,取出反应釜,将反应产物倒入烧杯中,得到碳量子点荧光探针的前驱液;
第二步是碳量子点荧光探针的制备:首先,用层析色谱法进行分离,把填充好的硅胶柱竖直安装在铁架台上,向上述前驱液的烧杯里中加入40~60ml配好比例的展开剂,用胶头滴管吸取的前驱液,沿着柱壁以2ml/min速度滴加前驱液,结束后,再沿着柱壁加入以2ml/min的速度滴加20~40ml配好比例的淋洗剂,将层析分离后的产物再用透析袋透析,保留液用10~30ml去离子水稀释,得到表面含有羟基和羰基的碳量子点荧光探针。
实施例:利用间苯二胺为原料,用溶剂热法,可制备得碳量子点荧光探针,制备过程包括如下两个步骤。
第一步是碳量子点荧光探针的前驱液的制备:首先,准确的称取0.03g的前驱体溶解在40ml的无水乙醇中,溶解后,移入到体积为100ml的聚四氟乙烯内胆的反应釜中,用材质为聚四氟乙烯盖密闭,金属盖密封,将其置于程序升温速率为5℃/min的干燥箱中,升温至100℃的范围内反应1h后,再升温至160℃的范围内反应12h后,自然冷却至室温,取出反应釜,将反应产物倒入烧杯中,得到碳量子点荧光探针的前驱液;
第二步是碳量子点荧光探针的制备:首先,用层析色谱法进行分离,把填充好的硅胶柱竖直安装在铁架台上,向上述前驱液的烧杯里中加入50ml配好比例的展开剂,用胶头滴管吸取的前驱液,沿着柱壁以2ml/min速度滴加前驱液,结束后,再沿着柱壁加入以2ml/min的速度滴加30ml配好比例的淋洗剂,将层析分离后的产物再用透析袋透析,保留液用20ml去离子水稀释,得到表面含有羟基和羰基的碳量子点荧光探针。
- 还没有人留言评论。精彩留言会获得点赞!