用于粉末涂漆的涂覆有机粘合剂的效应颜料、用于生产所述涂覆的效应颜料的方法及其用途与流程
本申请是由埃卡特有限公司于2017年3月30日提交的国际申请号为pct/ep2017/000392的发明名称为“用于粉末涂漆的涂覆有有机粘合剂的效应颜料、以及用于生产所述涂覆的效应颜料的方法及其用途”的国际申请的分案申请。该国际申请pct/ep2017/000392进入中国国家阶段的日期为2018年9月19日,国家申请号为201780018433.1。本发明涉及通过自发沉淀用有机粘合剂涂覆效应颜料,并且还涉及一种用于生产这些涂覆的效应颜料的方法及其用途。
背景技术:
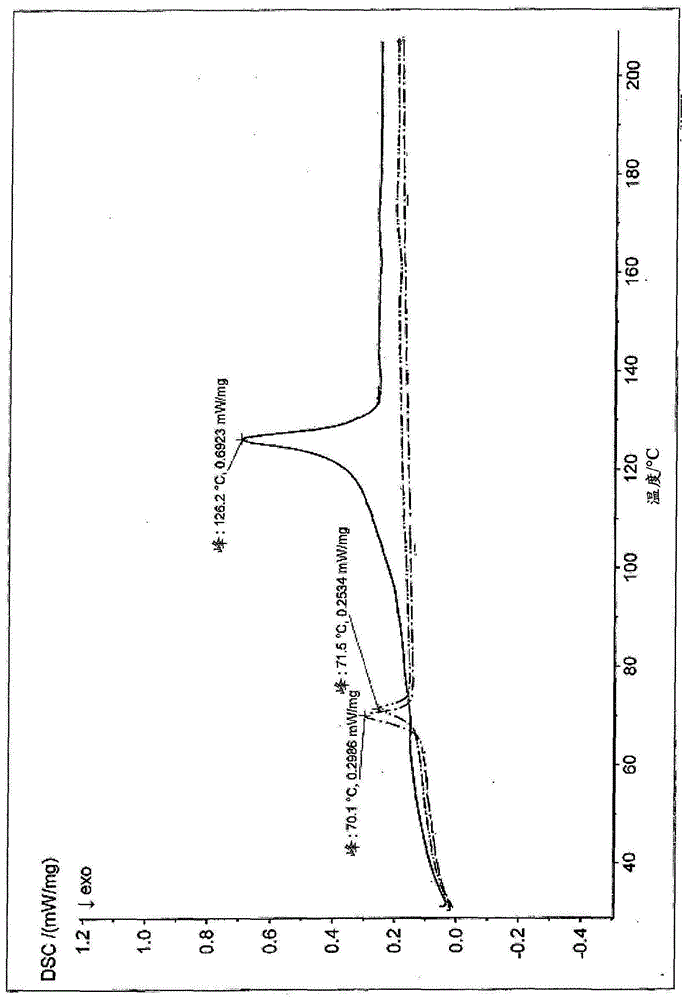
:效应颜料广泛用于涂漆(paints)、清漆(varnishes)、粉末涂料、印刷油墨、塑料或化妆品的着色。将这些颜料引入到粘合剂体系中并且润湿引起很多问题,特别是在粉末涂料的情况下。效应颜料不能像在有机或无机彩色颜料的情况下那样以通常的方式(通过挤出并且随后将挤出物粉碎成粉末涂料)结合入,因为这些操作会压碎片状颜料并且使它们失去其光学效应。相反,使用被称为干混或粘结工艺的工艺。干混方法是一种简单的混合操作,其中将商业粉末涂料和效应颜料彼此干燥结合。任选地,有可能将另外的添加剂例如像流动助剂加入混合操作中。缺点是,由于包括不同比重和静电充电行为的原因,这种类型的干混合物导致在施用粉末涂料期间效应颜料和粘合剂的分离。粉末涂料的再循环能力就其本身而言是粉末涂料体系的巨大优势之一,但这在用效应颜料着色并且通过该方法生产的粉末涂料的情况下不再是可能的。粘结方法是商业粉末涂料与效应颜料的混合操作,其中通过加热该混合物实现效应颜料颗粒物理附接到粉末涂料颗粒上。因此,对于粘结过程,存在效应颜料到粉末涂料颗粒表面上的粘附。粘结的效应颜料具有相对较低的色调变化,因为现在在施用期间粉末涂料与效应颜料的分离大大减少。然而,一个缺点是粘结过程是非常“技术”的过程,需要大量的经验和专门技能。例如,反应器中局部过热的情况是有可能的,并且然后导致在粉末涂料的一部分上不希望的硬化反应。然后必须花费成本且不方便地除去在反应器中形成的沉积物。如果没有通过工艺下游的保护性筛选完全去除沉积物,则可能在涂覆材料中形成小块(bit)。此外,在粘结过程中,取决于颗粒尺寸,仅有可能引入最多约7wt%的效应颜料。特别是在相对大的颜料(d50>35μm)的情况下,这些少量通常不足以实现足够的遮盖力。对于干混和粘结方法二者,缺点是效应颜料不完全被粘合剂包封并且因此在没有粘合剂包封的情况下施用到基材上。在随后的固化程序(通常是烘烤操作)过程中,用粘合剂润湿这些颜料是不完全的。其结果是,例如,在粉末涂料固化后金属效应颜料没有被粘合剂完全包围,并且因此它们也没有经受最佳的防腐蚀保护。粘结的效应颜料的另一个缺点是在一些情况下它们具有有限的保质期。鉴于这些粘合剂原则上是反应性的,这些产品可能由于在延长的停工时间期间粘合剂交联的部分开始而经受附聚,特别是在夏季或在炎热气候中的相对高温下,并且因此可能不再是可使用的。在ep1699884b1和ep1893699b1中提出了粘结产品的一种替代方案。在其中,通过喷雾干燥将反应性粘合剂与相应固化剂一起施用到片状金属效应颜料或珠光效应颜料上。为此目的,这些组分必须预先溶解在合适的有机溶剂中。粘合剂-固化剂体系在涂覆条件下不固化。相反,仅在干混操作之后与粉末涂料一起进行固化。这些产品具有粘结产品的优点,并且还有在金属效应颜料的情况下,与所使用的金属效应颜料相比,在相关化学品试验和砂浆试验中具有改进的耐受性,并且还具有特定的光学品质(3d效果)(u.hirth,a.albrecht和b.schreiber,1ppcj2008年12月)。然而,这些产品受制于相对较低的保质期。另外,在这些颜料的生产中,生产程序的成本和不便意味着以相对高的生产成本仅可以生产有限的量。因此需要涂覆的效应颜料,其与粘结产品一样具有效应颜料与粉末涂料的减少的分离,并且相对于现有技术具有改善的保质期。de102008060228a1或us7,578,879b2描述了涂覆有lcst和ucst聚合物的效应颜料。lcst聚合物是在低温下可溶于溶剂并且在限定的温度之上变得不可溶的聚合物。ucst聚合物是在高温下可溶于溶剂并且在低于某一温度下变得不可溶的聚合物。然而,这些聚合物是已经经过固化的聚合物,并且不是粘合剂。此外,这些文献的传授内容包括用于将聚合物固定在效应颜料表面上的后续反应。这些颜料不具有合适的遮盖力,并且倾向于附聚。在ep1332182b1中,描述了通过添加聚合物不溶于其中的溶剂将聚合物沉淀到颜料上,这些颜料来自颜料与聚合物在聚合物可溶于其中的溶剂中的分散体。在de2603211中,披露了相反的过程:将颜料或珠光颜料和聚合物在具有对聚合物的良好溶解性的溶剂中的分散体加入聚合物在其中具有差溶解性的溶剂中。此外,在de1544830中披露了一种在塑料中使用颜料浓缩物的相似方法。然而,这些方法不使结晶聚合物层沉淀。此外,在实施这些沉淀过程时,形成高度附聚的沉淀物,这使得持续加工几乎不可能。在效应颜料的情况下,获得了不太适合于施用的粘性粉末。还已知的是效应颜料、尤其是金属效应颜料,其涂覆有聚合物层,这些聚合物层由液相中的单体原位聚合到效应颜料上。大多数已知体系是或包含聚丙烯酸酯(ep2479224a1、us20120065298a1、ep2115075a1、ep1950257a1、ep1837380a1、ep477433b11)。在用聚丙烯酸酯涂覆之前,这些颜料中的一些涂覆有金属氧化物层(us20090117281a1、ep2688962a1、ep2773704a1)。在这些情况下,也总是使用多官能丙烯酸酯单体(交联剂)。因此形成的聚合物层是非晶的。这些产品在粉末涂料中表现出明显良好的可充电性和通常高的化学稳定性。然而,以这种方式涂覆的颜料通常以干混方法用于粉末涂料中,并且因此具有相应的缺点,如过度分离。在粘结过程中使用是不合适的,因为额外的聚合步骤使得该过程不必要地复杂并且昂贵。因此需要提供不具有所述缺点的涂覆的效应颜料。特别地,效应颜料具有非常好的保质期并且表现出粘结颜料的低分离倾向。此外,需要一种简单且廉价的用于生产这些颜料的方法。技术实现要素:本发明的目的是通过提供一种涂覆的效应颜料来实现的,该涂覆的效应颜料包含片状基材和施用在其上并且包含用于粉末涂覆材料的粘合剂的涂料;其特征在于,该粘合剂具有借助于13cnmrmas弛豫测量所确定的结晶和无定形部分,13c核的弛豫根据下式拟合为双指数弛豫其中结晶度c是在40%与85%之间的范围内,并且其中存在短平均弛豫时间t1s和长平均弛豫时间t1l,并且其中t1l是在从65至130s的范围内。该涂覆的效应颜料的另外的实施方案描述于从属权利要求2至14中。在方面2至18和方面37至57中还描述了另外的实施方案。此外,该目的通过提供一种用于生产涂覆的效应颜料的方法来实现,该效应颜料包含片状基材和施用在其上并且包含用于粉末涂覆材料的粘合剂的涂料;其特征在于,该方法包括以下步骤:a1)在温度tsol下在第一时间间隔tsol内将可自发沉淀的粘合剂溶解在有机溶剂或溶剂混合物中,b1)随后将效应颜料加入来自a1)的溶剂或溶剂混合物中,同时分散该效应颜料,c1)在温度tinsol下在第二时间间隔tinsol内用该粘合剂涂覆该效应颜料,或者a2)将效应颜料分散在溶剂或溶剂混合物中,b2)随后向具有温度tsol的该溶剂或溶剂混合物中加入可自发沉淀的粘合剂,c2)在温度tinsol下在时间间隔tinsol内用粘合剂涂覆该效应颜料,d)从该溶剂或溶剂混合物中去除该涂覆的效应颜料,并且e)任选地干燥该涂覆的效应颜料。该方法的其他实施方案描述于从属权利要求16至21中。在方面19至34中还描述了该方法的其他实施方案。i聚合物的自发沉淀:本发明是基于完全出乎意料的效果。该效果是聚合物最初在限定的温度tsol下溶解在有机溶剂中。通常通过诸如搅拌和/或超声处理的辅助措施来支持溶解。但是,在一定时间间隔tinsol之后,在溶液中形成聚合物的沉淀物。该过程自发发生,换言之,无需改变外部参数,例如像温度、ph、或由于加入盐或另一种溶剂。在本发明的上下文中,此行为被称为聚合物的“自发沉淀”。据本发明人所知,迄今为止还没有披露过聚合物的这种行为。根据本发明,还理解为自发沉淀的是由于例如释放溶剂化热的结果温度稍微变化时。在此轻微的温度变化应理解为在0℃至5℃、优选0.5℃至2℃范围内的变化。当然,在现有技术中长期建立的是作为温度变化、ph变化或通过添加盐(盐析)的结果,溶解的聚合物的沉淀。还已知通过向聚合物溶液中添加其中聚合物具有差溶解性的溶剂进行沉淀。例如,在ep1332182b1中,使用后一种方法来用有机聚合物涂覆颜料。另外已知的是ucst或lcst聚合物,然而,为了产生沉淀,温度变化总是强制性的。在本发明的上下文中利用了自发沉淀的效果,以便用有机粘合剂涂覆效应颜料。粘合剂优选是适用于粉末涂料的粘合剂。特别优选,用于此目的的粘合剂是粉末涂料粘合剂。在特别优选的实施方案中,所使用的粘合剂是在温度tinsol下在有机溶剂中可自发沉淀的粘合剂。在合适的实施方案中,时间间隔tinsol是在从约半分钟至48小时的范围内。在该时间间隔内,沉淀至少50%、优选60%、更优选70%、非常优选至少80%、并且特别优选至少90%的先前溶解的聚合物。然而,低于半分钟的时间间隔不适合涂覆效应颜料,因为沉淀是在不充分控制下进行。对于超过48h的时间,由于长批次时间的成本原因,沉淀过程是不利的。因此,优选地,在来自从0.5h至12h范围并且更特别优选来自从1h至10h范围、并且尤其优选来自从2h至8h范围的时间间隔tinsol内在有机溶剂中沉淀至少50%、优选60%、更优选70%、非常优选至少80%、并且特别优选至少90%的先前溶解的聚合物。非常特别优选地,至少90%的粘合剂在来自从2至8h范围的时间间隔tinsol内沉淀在有机溶剂中。总体上,已观察到聚合物的沉淀在很大程度上是定量的。总体上,90%至100%、优选92%至97%的溶解的聚合物再次沉淀。自发沉淀的温度tinsol优选是在从0℃至80℃的范围内、更优选在从10℃至70℃的范围内、更特别优选在从15℃至50℃的范围内、并且尤其优选在从18℃至30℃的范围内。在较低温度下比在较高温度下更快地发生沉淀。用于溶解的溶剂不应该过于非极性。因此,在非极性溶剂例如像四氯化碳中,已经观察到粘合剂可以溶解,但它们不再次沉淀。优选的有机溶剂是来自酮和酯的组的溶剂。特别优选的溶剂是丙酮、甲基乙基酮(mek)、乙酸乙酯、以及其混合物。虽然在自发沉淀之前聚合物的溶解确实发生而没有可见的残余物,但溶液总是具有一定的不透明性,表明由于溶解的聚合物的聚集而存在胶体状态。相反,在例如其中没有发生自发沉淀的非极性溶剂如四氯化碳中,得到澄清的溶液。在优选的实施方案中,粘合剂具有聚酯官能团。在特别优选的实施方案中,粘合剂是聚酯,其可以用作粉末涂料粘合剂。这种粘合剂通过熔融聚合制备。在这种情况下,使单体(作为酸组分含有羧酸官能团的单体和作为醇组分含有醇官能团的单体)达到熔融状态,并且在合适的催化剂存在下通过缩聚进行低聚或聚合。形成聚酯,其可以具有羟基或羧酸基团作为末端官能团。这些聚酯当然不是完全固化的,因为实际上,当烘烤涂料时,它们需要在涂覆操作后在粉末涂料中作为粘合剂被进一步固化。与制备此类聚酯有关的实例在ep0521992b1、us6,660,398b1、us6,635,721b1、wo2001/138432a1、us6,384,102b1、wo2004/083326a1、和wo2002/50201a1中找到。在另外优选的实施方案中,聚酯包含由间苯二甲酸和对苯二甲酸及其混合物组成的组的单体作为酸组分的主要部分。在特别优选的实施方案中,该粘合剂包含基于所有的酸组分的至少14mol%的间苯二甲酸作为酸组分。基于所有酸组分,间苯二甲酸和对苯二甲酸的分数优选是50至100mol%并且更优选70至96mol%。优选不作为主要部分包括的另外的酸组分可以是己二酸、邻苯二甲酸、琥珀酸、戊二酸、庚二酸、壬二酸、癸二酸、富马酸、马来酸、1,3-环己烷二羧酸、1,4-环己烷二羧酸、以及所呈现的这些化合物的混合物。还作为支化单元以少量(低于10mol%)存在的可以是具有三个或更多官能度的羧酸,如偏苯三酸或均苯四酸。关于聚酯粘合剂的醇组分,优选的实施方案是以新戊二醇和/或2-丁基-2-乙基-1,3-丙二醇作为醇组分的主要部分为特征的那些。新戊二醇和/或2-丁基-2-乙基-1,3-丙二醇的分数优选为基于在所有醇组分的50至100mol%、更优选70至98mol%、并且非常优选80至95mol%。在此非常特别优选新戊二醇是主要的醇组分。具有这种醇含量的聚酯粘合剂倾向于在文献中被归属为“无定形”聚酯(us6,660,398b1、us6635721b1)。作为另外的醇组分,优选总计小于所有醇组分的50mol%可以是例如乙二醇、1,3-丙二醇、1,4-丁二醇、1,6-己二醇、2-甲基-1,3-丙二醇、1,4-环己烷二甲醇、氢化双酚a、新戊二醇羟基新戊酸酯、以及所呈现的这些化合物的混合物。作为支化单元还以少量(低于10mol%)存在的可以是含有至少三个羟基的具有三个或更多官能度的羟基化化合物,例如像三羟甲基丙烷、双三羟甲基丙烷、季戊四醇、及其混合物。在进一步改进的实施方案中,关于聚酯的醇组分,直链α,ω-醇(例如像乙二醇)与新戊二醇和/或2-丁基-2-乙基-1,3-丙二醇的摩尔比率是低于20%、更优选低于15%、甚至更优选低于12%、并且非常优选低于10%。具有过高水平的直链α,ω-醇的聚酯(例如像乙二醇)已被证明不太适合自发沉淀。这些种类的聚酯在文献中被称为更“无定形的”聚酯。具有高比例的更多对称单体的聚酯,例如像直链α,ω-醇和/或具有高分数的对苯二甲酸,在文献中被称为“半结晶”聚合物(ep0521992b1、us4,217,426)。聚合物的自发沉淀可归因于在聚合物溶解在溶剂中之后开始的聚合物结晶增加。下面将使用各种测量方法表现该事实。不受理论束缚,特别是在适用于粉末涂料的聚酯粘合剂的情况下,对此的解释是,这些聚酯虽然它们可能确实已经具有某些部分结晶度,但是由于它们由熔体制备而具有非常大的空间应变。作为溶解的结果,聚合物链需要更大的运动自由度,并且合适的聚合物链段有可能彼此找到并且形成结晶区域。因此,最终,该聚合物变得不可溶并且沉淀。值得注意地并且完全出乎意料地,这在无需进一步改变外部参数的情况下发生。在具有高分数的新戊二醇作为醇组分和/或具有高分数的间苯二甲酸作为酸组分的聚酯的情况下,这是尤其显著,因为这些粘合剂特别在文献中被描述为无定形聚酯(us6,660,398b1、ep0521992b1)。ii聚合物的表征iiadsc测量:差示扫描量热法(dsc)是用于分析聚合物的标准方法。基于吸热熔融峰的比能量,可以估计结晶度。然而,为了确定精确的结晶度,总是需要基于具有类似结构的已知聚合物的标准化。在本发明中主要通过温度最大值tmax确定熔融峰的位置。根据本发明沉淀的聚合物也可以具有带有多个最大值的熔融峰。在这种情况下,对于最高峰选择tmax;如果两个峰具有大致相同的强度(+/-5%),则取两个最大值的平均值。涂覆有根据本发明的粘合剂的效应颜料表现出在5℃/min前进速率下的dsc图中优选至少一个吸热峰,其具有来自tmax=100℃至150℃的范围的最大值,并且还具有与该峰值相关来自从15j/g至80j/g范围的焓δh,该焓是根据粘合剂的量计算的。该吸热峰归属于粘合剂的结晶部分的熔化。进一步优选地,tmax位于从110℃至140℃的范围内。此外,优选地,熔化焓位于20-70j/g的范围内,并且更优选在30-65j/g的范围内,该焓根据该粘合剂的量进行计算。在聚酯粘合剂的情况下,熔化焓的位置tmax是显著的。如果对未沉淀的粘合剂进行dsc,则在优选的聚酯粘合剂的情况下,第一次dsc操作包括具有约65℃至72℃的tmax的非常小的峰。该峰可能很大程度上归因于聚合物的某些小部分结晶区域。因此,根据本发明的涂覆有粘合剂的效应颜料在dsc中表现出与未结晶状态相比来自从40℃至75℃范围并且优选来自从50℃至70℃范围的结晶状态的熔化焓增加(δtmax)。未沉淀的粘合剂、优选聚酯粘合剂的熔化焓(δhagm)水平在仅约5至10j/g的范围内。因此,根据本发明涂覆有粘合剂的效应颜料具有在从2至10的范围内的吸热熔化焓的比率δh/δhagm,其可以通过以5℃/min的前进速率进行dsc测量来确定,并且其基于沉淀后(δh)的粘合剂的量相对于沉淀前(δhagm)的粘合剂。优选地,该比率是在从3至8的范围内,并且尤其优选在从4至7.5的范围内。iib粉末衍射(pxrd):德拜-谢勒(debye-scherrer)几何中的粉末衍射法是确定聚合物结晶度的常规方法。先决条件是样品在空间上以统计分布存在,并且尽可能没有优先方向。该方法仅可以用于检测高于一定尺寸的结晶部分,因为如果微晶太小则不存在衍射。pxrd衍射图优选使用cukα1作为x射线源并且使用具有0.5mm的狭缝宽度的锗(111)作为单色器以德拜-谢勒几何(毛细管直径0.3mm)进行记录。所有这些参数影响所得峰的宽度。根据本发明涂覆的效应颜料在此具有结构化衍射图。在这种情况下的峰可以归属于(半)结晶粘合剂。这些峰具有在从0.7°至2.0°(以2θ计)的范围内的半峰全宽。相反,未沉淀的聚合物不产生结构化光谱。iic13cmasnmr光谱和弛豫:因为固体的nmr弛豫测量虽然在用于表征聚合物的结晶和无定形部分的科学文献中是众所周知的,但是在专利文献中较少提及,首先给出该方法的一般描述。mas光谱学:对于固态nmr光谱学,影响光谱的强核-核相互作用的影响是特有的。在液态nmr中,这些相互作用主要通过布朗运动达到平均。这导致窄的共振谱线,从中可以得到关于待分析样品的化学组成的深入信息。对于其中存在固体结构的凝聚相(固态)材料,布朗运动较弱或完全不存在。因此,自旋的相互作用总是存在,这导致无结构的、非常宽的nmr光谱。在这些相互作用中,偶极相互作用、化学位移各向异性和四极相互作用起到最大作用。鉴于四极相互作用仅与具有核自旋i>1的核相关,在13cnmr的情况下可以忽略它们(类似于1h,对于13c,自旋=1/2)。化学位移各向异性(简称csa)描述了各向同性与各向异性状态之间nmr信号的化学位移差异。已知化学位移是特定原子核的局部磁场的量度,从而产生确定的磁矩。在液态nmr中,由于聚合物化学键周围构象的快速变化,聚合物的化学位移通常是所有可能构象的平均值。在固体中,化学位移(即,核自旋与分子轨道中的电子之间的相互作用)受到任何结构变化的影响。因此,它不仅含有关于化学特性(identity)的有价值的信息,而且还含有关于所涉及的分子基团的分子取向的有价值的信息。偶极相互作用,也称为偶极耦合,描述了双自旋与磁矩的直接相互作用。在13c核和与其结合的质子(1h)的情况下,偶极耦合取决于距离r(~1/r3)和其相对于外部磁场b0的取向(θ)[isaoando,tetsuoasakura,solidstatenmrofpolymers[聚合物的固态nmr],第84卷]。为了减少这些影响,andrew等人于1958年提出了一种新方法[e.r.andrew,a.brandbury,r.g.eades,nature[自然]1959,183,1802.]。这种新技术(用于幻角自旋的mas)涉及测量以特定角度θm(“幻”角)旋转的陶瓷转子中的样品,旋转频率νrot为~1khz至70khz。幻角θm是旋转轴与外部磁场b0之间的角度,其中表示平均空间取向的项,cos2θm=1/3是成立的。精确的角度是54.74°。在这个角度,拓宽nmr线的自旋相互作用部分地被平均,结果线宽明显减小。然而,该宽度仍然是液态nmr中相应信号的线宽的约10至100倍。在几何术语中,幻角可以被描述为立方体的对角线与边的角度。masnmr光谱的特征是在各向同性化学位移的位置处存在中心线,并且还存在边带(被称为“旋转边带”(ssb))。旋转边带(ssb)总是出现在旋转频率νrot的整数倍处。它们在光谱中总是标有上标星号(*)。在具有多个共振的聚合物样品的情况下,旋转边带与各种化学基团的化学位移的实际信号的重叠使得评估记录的光谱更加困难。因此,通常使用其中至少相关化学基团的中心线没有重叠的旋转频率。在这种情况下,通常,较高的旋转频率将旋转边带从光谱中去除。对于具有大的化学各向异性位移的基团(例如,对于具有coo基团的聚合物),使用高旋转频率和/或用于抑制旋转边带的特定技术[w.t.dixon,j.chem.phys.[物理化学杂志],77,1800(1982);o.n.antzutkin,z.song,x.feng,和m.h.levitt.j.chem.phys.[物理化学杂志],100,130(1994)]。通常,几khz直到20khz的旋转频率足以平均13c核与键合到其上的质子之间的偶极相互作用。1h核彼此之间的偶极相互作用甚至更强,并且因此必须使用高达70khz的更高旋转频率。因此,1hmas固态nmr需要极高的成本和材料的复杂性,并且因此很少使用。在聚合物的情况下,13cmasnmr在一段相当长的时间内出现,其足够敏感以分析结构和动态过程。由于13c核具有比1h核低得多的固有频率,因此常规技术涉及将更常见的物种(因此在聚合物的情况下,通常为1h)磁化为稀有物种(13c)。该领域最常见的技术之一是hartmann-hahn交叉极化,其中激发的质子核的磁化被转移到13c核。在这种情况下,如所涉及的核中的旋磁比(gyromagneticratio)(γh/γc)所给出的,作为灵敏度的最大增加可以达到因子4[s.r.hartmann,e.l.hahn,phys.rev.[物理评论]1962,128,2042]。在晶体中,分子以不等价的位置或冷冻构象存在。在此,固态masnmr光谱将具有更多化学位移稍微改变的信号,而在溶液态nmr中,信号将是等效的。这种效应可以用于固态nmr以分析互变异构及其动态变化[f.laupretre,l.monnerie,j.virlet.macromolecules[大分子]17(1984)1397]。对于低于tg的无定形聚合物样品,相对较大的链无序将带来比在结晶样品情况下更宽的nmr带。无定形相的宽nmr信号与结晶相的较窄信号之间的差异可以用于确定聚合物的聚合度[isaoando,tetsuoasakura,solidstatenmrofpolymers[聚合物的固态nmr],第84卷]。鉴于聚合物的无定形相与结晶相中的链构象不同,其nmr光谱和其动态nmr行为也将是不同的。对于半结晶聚合物,动态行为可以用作具有高度可移动链的无定形相的行为与其中链的运动自由度受到很大限制的结晶相的行为之间进行区分的标准。强烈使用的方法之一是t1纵向弛豫技术[w.s.veeman.e.m.menger.h.h.c.demoor,proc.iupaci.u.p.a.c.macromol.svmp.[大分子研讨会文集]28rh.1982.2;f.c.schilling,f.a.bovery,a.e.tonelli.polvm.marer.sci.eng.50(1984)256,d.a.torchia.j.magn.reson.[磁共振杂志]30(1978)613.]。13cmasnmr弛豫:纵向弛豫或自旋晶格弛豫是描述自旋体系中磁化矢量的发展直至达到平衡的机制,该平衡在磁化矢量与外场bo平行时获得。通过合适的90x`°脉冲,将通过定义在平衡态中在z方向(平行于外部磁场)上指向的旋转坐标系中的宏观磁化m0移动到y`-方向。能量水平的占有率发生变化,因为发生了等占用(equioccupancy),而不是根据在静止状态(stateofrest)下应用的玻尔兹曼分布的占用。随后,根据具有时间常数t1的指数函数,使弛豫成为平衡状态。由于链在无定形相中的高迁移率,t1显著短于结晶相中的弛豫时间。这种效果的结果是,在半结晶聚合物的情况下,弛豫曲线不再能通过以下类型的简单指数函数来描述[blochf.phys.rev.[物理评论]1946;70:460-474.]:相反,在此有必要使用双指数函数,以便将无定形部分的快速弛豫时间t1s与结晶部分的缓慢弛豫时间t1l分开:在此,以下指配是有效的:m0:平衡磁化m(t,m0,a,c,t1s,t1l):在时间t下的磁化c:以摩尔分数计的结晶聚合物的比例t:90°脉冲后的时间a:该参数考虑了非零初始磁化,并且与确定聚合物和涂覆的al颗粒中的结晶部分无关。参数a反映饱和区段(block)和基线校正的效率,并且在t=0时完全消除磁化的情况下应该接近1.0。t1s:自旋晶格弛豫时间“短”:无定形部分的弛豫时间t1l:自旋晶格弛豫时间“长”:结晶部分的弛豫在90°脉冲之间插入合适的等待时间。总之,该方法对于检测聚合物的结晶部分非常敏感。因为该方法必须始终在分子上解释,所以甚至可以检测聚合物中非常小的结晶域。iid对聚合物和本发明涂覆的效应颜料进行13cnmrmas测量有待进行的测量的实验细节在实验部分中找到。根据本发明的涂覆的效应颜料的特征在于,该粘合剂具有借助于13cnmrmas弛豫测量来确定的结晶和无定形部分,13c核的弛豫根据下式拟合为双指数弛豫其中结晶度c是在40%与85%之间的范围内,并且其中存在短平均弛豫时间t1s和长平均弛豫时间t1l,并且其中t1l是在从65至130s的范围内。在其他实施方案中,该粘合剂具有45%至75%的结晶度c。在优选的实施方案中,长平均弛豫时间t1l是在从70至110s的范围内、更优选在从71至100s的范围内。iii效应颜料:这些效应颜料取自由金属效应颜料、珠光颜料和干涉颜料、以及其混合物组成的组。金属效应颜料是基于片状金属芯。在此被称为片状的金属芯是其纵横比(即,平均尺寸d50与平均厚度h50的比率)是大于5的芯。进一步优选的是具有10至500和40至100的纵横比的金属芯。具有更高纵横比的金属芯具有更好的金属特性,如浅黑色随角异色(flop)或光泽。根据本发明,优选片状金属芯包含来自下组的金属或由其组成,该组由以下各项组成:铝、铜、锌、锡、金青铜(goldbronze)、黄铜、铁、钛、铬、镍、银、金、钢以及其合金及混合物。根据本发明的另一变体,片状金属芯是铝颜料或金青铜颜料。铝颜料是特别优选的。金属效应颜料是片状的和基本上圆形的(被称为“银元”的类型),或片状和基本上“玉米片状”(被称为“玉米片”的类型)。本发明的金属效应颜料的片状金属芯(即,没有随后的聚合物涂覆)优选具有约20nm至约2μm、更优选25nm至1μm、并且还更优选地为30nm至600nm的平均厚度h50。h50值涉及厚度分布的累积频率分布。厚度分布可以借助于对于wo2004/087816a2(第24和25页)中描述的方法中的第70至100颜料的sem测量来确定。本发明的效应颜料的尺寸通常借助于激光粒度测量来确定。此分析产生体积平均尺寸分布函数的累积频率分布。在这种情况下,d50值表示50%的所测量效应颜料具有等于或小于所指出的特定值的体积平均直径所处于的值。根据fraunhofer理论评估散射光信号,该理论还包括颗粒的折射和吸附行为。尺寸分布优选使用来自康塔公司(quantachrome)的cilas1064仪器进行测量。本发明的效应颜料的尺寸分布的d50值优选是在从3至200μm范围内、更优选在从5至100μm范围内、并且非常优选在从8至90μm范围内。在另外的实施方案中,该片状基材是涂覆有金属氧化物的金属效应颜料,或者是珠光颜料。在此的金属氧化物优选取自由(二)氧化硅、氧化硅水合物、氢氧化硅、氧化铝、氧化铝水合物、氢氧化铝、及其混合物组成的组。特别优选二氧化硅(sio2)。以这种方式用金属氧化物预涂覆的金属效应颜料本质上更容易根据本发明用适合于粉末涂覆的粘合剂进行涂覆。此外,就整体效应颜料而言,金属氧化物涂料有助于改进的化学稳定性和/或气体处理稳定性。金属氧化物优选具有至少10nm的几何层厚度。在此层厚度之下,化学稳定性将不足。金属氧化物的优选最大层厚度是200nm,优选50nm。在200nm之上的层厚度下,效应颜料的光学特性可能变得不利。在一个特别优选的实施方案中,片状基材是sio2涂覆的铝效应颜料。在根据本发明的发展中,该金属颜料可以用增强效应颜料表面与沉淀到其上的粉末涂料树脂之间的粘附性的物质进行预涂覆。此物质的可能实例包括官能化硅烷、官能化聚合物和有机磷化合物。优选使用官能化硅烷。这些硅烷优选具有通式(iii)(y)r(4-z)si(x)z(iii)在具有式(iii)的硅烷化合物中,z是从1至3的整数,r是具有1至12个c原子的取代或未取代的、未支化或支化的烷基链,y是能够与沉淀聚合物的相应的官能团反应的官能团,并且x是卤素和/或烷氧基。r也可以环状地与si连接,在这种情况下,z通常为2。x优选为甲氧基或乙氧基。基团y可以在末端与烷基r键合。两个或更多个基团y也可以与一个烷基键合。在si(x)结构部分与金属颜料表面的表面oh基团缩合反应之后,硅烷与此表面结合。相反,反应性官能团y可以附接到随后沉淀在其上的粉末涂料粘合剂上。这种附接可能涉及共价键或不太强的相互作用,如氢键。关键因素是,借助于作为粘合促进剂的硅烷,粉末涂料粘合剂锚定在金属颜料表面上,其强度使其在随后例如通过筛分的效应颜料的进一步加工或进一步加工成粉末涂料的过程中保持大部分粘接在效应颜料上。因此,硅烷充当金属颜料表面与涂料的粉末涂料粘合剂之间的粘合促进剂。优选的官能团y是异氰酸酯、环氧基、氨基、羟基、羧基、丙烯酸酯或甲基丙烯酸酯基团。这些基团与粉末涂料粘合剂的相应的化学相容的对应基团反应。然而,在此的粘合剂就其本身而言不固化;换言之,低聚物/聚合物粘合剂保持其可化学交联性或可固化性。硅烷的官能团y可以与例如不参与或仅部分参与粉末涂料粘合剂的固化的粉末涂料粘合剂的官能团反应。例如,基于硅烷的官能团y的粉末涂料粘合剂的官能团可以化学计量过量存在。在用至少一种具有式(iii)的硅烷化合物预涂覆的金属颜料表面上,基于随后施用的粉末涂料粘合剂的相应的化学相容的官能对应基团,官能团(y)通常是化学计量不足的。例如,y可以是异氰酸酯基团或环氧基团,而粘合剂包含具有多元醇和多羧基官能团的聚酯组分。在适当时加入催化剂,异氰酸酯基团甚至能够在室温下与粘合剂的oh基团反应。然后,聚酯涂料在涂料体系的烘烤过程中仅在金属颜料被涂覆并掺入到涂料体系中之后固化。基团y优选为端基,因为在这种情况下由于空间位阻最小反应性最大。可替代地,它可以是大端基,其中在y官能团之前可能另外存在到链末端上的高达3个c原子。与y反应的粘合剂官能团也可以与在粘合剂固化时构成聚合物的那些相同。这是可能的,因为如以上已经观察到的粉末涂料粘合剂的y-反应性官能团通常以相对颜料表面上的官能团y的化学计量过量存在,并且因此,在反应性基团y已经与低聚物/聚合物粘合剂反应之后,在低聚物和/或聚合物粘合剂上仍存在足够的官能团用于交联或固化。与反应性基团y是反应性的粉末涂料粘合剂的官能团也可以与参与粘合剂固化的一个或多个官能团不同。具有相应官能团的适合作为表面改性剂的有机官能硅烷是可商购的。例如,存在莱茵费尔登的赢创公司(evonik)生产并以商品名出售的产品、或由特种有机硅公司(osispecialties)生产的硅烷、或由瓦克公司(wacker)生产的硅烷的许多代表。其实例是3-甲基丙烯酰氧丙基三甲氧基硅烷(dynasylanmemo,silquesta-174nt)、3-巯基丙基三(甲氧基)乙氧基硅烷(dynasylanmtmo或3201;silquesta-189)、3-缩水甘油基氧基丙基三甲氧基硅烷(dynasylanglymo,silquesta-187)、三(3-三甲氧基甲硅烷基丙基)异氰脲酸酯(silquesty-11597)、γ-巯基丙基三甲氧基硅烷(silquesta-189)、β(3,4-环氧基环己基)乙基三甲氧基硅烷(silquesta-186)、γ-异氰酸丙基三甲氧基硅烷(silquesta-link35,genosilgf40)、(甲基丙烯酰氧基甲基)三甲氧基硅烷(genosilxl33)、(异氰酸基甲基)三甲氧基硅烷(genosilxl43)、氨丙基三甲氧基硅烷(dynasylanammo;silquesta-1110)、氨丙基三乙氧基硅烷(dynasylanameo)或n-(2-氨乙基)-3-氨丙基三甲氧基硅烷(dynasylandamo,silquesta-1120)或n-(2-氨乙基)-3-氨丙基三乙氧基硅烷、三氨基官能三甲氧基硅烷(silquesta-1130)、双(γ-三甲氧基甲硅烷基丙基)胺(silquesta-1170)、n-乙基-γ-氨基异丁基三甲氧基硅烷(silquesta-link15)、n-苯基-γ-氨丙基三甲氧基硅烷(silquesty-9669)、4-氨基-3,3-二甲基丁基三甲氧基硅烷(silquesty-11637)、(n-环己基氨基甲基)三乙氧基硅烷(genosilxl926)、(n-苯基氨基甲基)三甲氧基硅烷(genosilxl973)、及其混合物。硅烷、优选具有式(i)的硅烷可以直接施用在效应颜料的金属表面上。根据一个优选的发展,效应颜料是具有sio2涂料、优选包封有sio2涂料的金属颜料,将硅烷施用在sio2涂料上。然后将粉末涂料粘合剂施用到如此预涂覆的金属颜料上。iv方法:一种用于生产本发明涂覆的效应颜料的方法包括以下步骤:a1)在来自从0℃至80℃的范围内的温度tsol下在tsol的第一时间间隔内将可自发沉淀的粘合剂溶解在有机溶剂或溶剂混合物中,b1)随后将效应颜料加入来自a1)的溶剂或溶剂混合物中,同时分散该效应颜料,c1)在来自范围从0℃至80℃的温度tinsol下在第二时间间隔tinsol内用该粘合剂涂覆该效应颜料,a2)将效应颜料分散在溶剂或溶剂混合物中,b2)随后向具有温度tsol的该溶剂或溶剂混合物中加入可自发沉淀的粘合剂,c2)在时间间隔tinsol内用粘合剂涂覆该效应颜料,d)从该溶剂或溶剂混合物中去除该涂覆的效应颜料,并且e)任选地干燥该涂覆的效应颜料。因此,存在两种用于生产本发明涂覆的效应颜料的主要方法。在第一种方法中,首先将可自发沉淀的粘合剂溶解在有机溶剂或溶剂混合物中。就其本身而言,时间间隔tsol不重要,并且将由实际考虑因素决定。优选地,时间间隔tsol是在从约0.25至3h的范围内。通过搅拌和/或超声处理该溶液有利地促进溶解。在此的粘合剂的浓度是基于溶剂的量在从约10至40wt%、优选15至25wt%的范围。为了节省溶剂,有可能在相对高的浓度下操作,但要记住效应颜料仍然可以有效地分散。第二步骤b1)(将效应颜料加入溶剂中并且将效应颜料分散在溶液中的步骤)应该有利地在粘合剂溶解结束后立即进行。如果在此经过的时间太长,则粘合剂的自发沉淀可能在此早期阶段开始。在加入溶解的聚合物之前,效应颜料可以已经采用在溶剂中、优选在已经用于步骤a1)的溶剂或溶剂混合物中的单独分散体的形式。以这种方式可以使添加更容易,例如,通过用通常用于此目的的设备计量加入。还有可能的是,以这种方式防止了在效应颜料粉末的情况下有可能的粉尘,并且添加在几乎没有残余物下进行。第二种方法甚至更简单,并且根据本发明是优选的。在此,依照步骤a2)的效应颜料已经分散在溶剂或溶剂混合物中。已经发现,在步骤a2)中,所添加的聚合物在溶剂或溶剂混合物中的溶解很大程度上不受已经分散的效应颜料的影响。在步骤c2)中到效应颜料上的沉淀几乎是无缝的。步骤b2)和c2)也可以在时间上重叠。这不一定是缺点,因为尚未溶解的聚合物在很大程度上不损害已溶解的聚合物的沉淀。步骤c1)或c2)中的时间间隔tinsol是在从0.5h至12h的范围内、并且更优选在从1h至10h的范围内,并且非常特别优选地,在从2h至8h的范围内。也可能是从0.5h至1.5h的时间间隔。聚合物的沉淀很大程度上是定量的,优选92%至100%。然而,已经发现,不一定所有的聚合物涂覆这些效应颜料;某一部分也可以以二次沉淀物的形式存在。然而,特别是在粉末涂料应用中,这不是缺点。然而,效应颜料被粘合剂非常均匀地并且在很大程度上定量地涂覆在其区域中。步骤c1)或c2)中的粘合剂沉淀温度tinsol优选是在从0℃至80℃的范围内、更优选在从比溶剂或溶剂混合物的沸点低10℃至5℃的范围内、进一步优选在从15℃至50℃的范围内、并且非常优选在从18℃至30℃的范围内。在较低温度下比在较高温度下更快地进行沉淀。温度有利地为室温或接近室温。这节省了与加热或冷却分散体的操作相关的能量消耗。步骤a1)或b2)的温度tsol也优选是在从0℃至80℃的范围内、更优选在从比溶剂或溶剂混合物的沸点低10℃至5℃的范围内、进一步优选在从15℃至50℃的范围内、并且非常优选在从18℃至30℃的范围内。在此同样是这样的情况:在室温或接近室温下操作提供了最大的优点。在本发明特别优选的方法中,两种方法的差δt=tinsol–tsol是在从0℃至5℃的范围内,并且因此存在自发沉淀。当然,在此该方法特别容易控制,因为在很大程度上可以没有加热或冷却步骤。然而,对于tsol或tinsol,也可以选择不同的温度。因此,例如,第一种方法中的粘合剂可以在较高温度下溶解,这通常比在较低温度下更快地进行,这之后可以在较低温度下进行沉淀。然而,对于tsol或tinsol具有不同温度的方法的变体不代表根据lcst或ucst聚合物的方法,因为本发明优选不使用lcst或ucst聚合物作为粘合剂。优选地,步骤c1)或c2)中的tinsol是在从0.5h至12h的范围内并且更优选在从2至8h的范围内。此外,还有可能自从1h至2h的范围内选择沉淀时间tinsol。如果tinsol低于0.5h,则沉淀可能不受控制,二次沉淀的分数太高,即,粘合剂不沉淀到效应颜料的表面上。如果tinsol是高于12h,那么在产品方面没有预期的缺点,但是固有地极其简单的方法变得不必要的长。有利地选择溶剂,其中沉淀时间tinsol短,在约1h至3h范围内,并且温度tinsol处于或接近室温,优选18℃至30℃。有待涂覆的效应颜料可以以糊剂或粉末的形式存在。作为其生产的结果,金属效应颜料通常呈含有涂料溶剂(whitespirit)并且具有约50至70wt%的固体分数的糊剂的形式。在这种情况下,将另一种溶剂引入相对非极性的体系中。这不需要不利地影响沉淀过程,但是应该使用具有相应溶剂混合物的预实验来确定所用粘合剂是否可以在这些体系中沉淀。效应颜料优选呈粉末形式。在此,一方面,聚合物沉淀的条件被更好地限定,但另一方面,关于溶剂后处理的可能性,如果不存在溶剂混合物则是有利的。对于这两种方法,优选地该有机溶剂选自由丙酮、甲基乙基酮、乙酸乙酯、以及其混合物组成的组。在此,有利地,决定将有利于一种溶剂而非混合物。当效应颜料是最初以含有涂料溶剂(whitespirit)的糊剂形式存在的金属效应颜料时,如以上被认为优选的溶剂将存在于与涂料溶剂(whitespirit)的混合物中。在这种情况下,应该确保存在于金属效应颜料糊剂中的涂料溶剂(whitespirit)或另一种非极性溶剂基于整体溶剂以优选低于20wt%、更优选低于10wt%的浓度存在于分散体中。这些方法的步骤d)和任选地e)是已知的标准步骤,并且可以以常规方式进行。总而言之,本发明的方法因其极其简单和低成本而众所周知。所使用的溶剂可以循环;换言之,在步骤d)之后分离出的溶剂可以返回(任选地在简单后处理之后返回)到涂覆过程中。在本发明的意义上,本发明方法中使用的粘合剂是可自发沉淀的。在优选的实施方案中,粘合剂具有聚酯官能团。在特别优选的实施方案中,粘合剂是聚酯,其可以用作粉末涂料粘合剂。这种粘合剂通过熔融聚合制备。在这种情况下,使单体(作为酸组分含有羧酸官能团的单体和作为醇组分含有醇官能团的单体)达到熔融状态,并且在合适的催化剂存在下通过缩聚进行低聚或聚合。形成聚酯,其可以具有羟基或羧酸基团作为末端官能团。这些聚酯当然不是完全固化的,因为实际上,当烘烤涂料时,它们需要在涂覆操作后在粉末涂料中作为粘合剂被进一步固化。在另外优选的实施方案中,聚酯包含作为酸组分的由间苯二甲酸和对苯二甲酸及其混合物组成的组的单体的主要部分。在特别优选的实施方案中,该粘合剂包含基于所有的酸组分的至少14mol%的间苯二甲酸作为酸组分。基于所有酸组分,间苯二甲酸和对苯二甲酸的分数优选是50至100mol%并且更优选70至96mol%。优选不作为主要部分包括的另外的酸组分可以是己二酸、邻苯二甲酸、琥珀酸、戊二酸、庚二酸、壬二酸、癸二酸、富马酸、马来酸、1,3-环己烷二羧酸、1,4-环己烷二羧酸、以及所呈现的这些化合物的混合物。还作为支化单元以少量(低于10mol%)存在的可以是具有三个或更多官能度的羧酸,如偏苯三酸或均苯四酸。关于聚酯粘合剂的醇组分,优选的实施方案是以新戊二醇和/或2-丁基-2-乙基-1,3-丙二醇作为醇组分的主要部分为特征的那些。新戊二醇和/或2-丁基-2-乙基-1,3-丙二醇的分数优选为基于所有醇组分的50至100mol%、更优选70至98mol%、并且非常优选80至95mol%。在此非常特别优选新戊二醇是主要的醇组分。具有这种醇含量的聚酯粘合剂倾向于在文献中被归属为“无定形”聚酯(us6,660,398b1、us6635721b1)。作为另外的醇组分,优选总计小于所有醇组分的50mol%可以是例如乙二醇、1,3-丙二醇、1,4-丁二醇、1,6-己二醇、2-甲基-1,3-丙二醇、1,4-环己烷二甲醇、氢化双酚a、新戊二醇羟基新戊酸酯、以及所呈现的这些化合物的混合物。作为支化单元还以少量(低于10mol%)存在的可以是含有至少三个羟基的具有三个或更多官能度的羟基化化合物,例如像三羟甲基丙烷、双三羟甲基丙烷、季戊四醇、及其混合物。在进一步改进的实施方案中,关于聚酯的醇组分,直链α,ω-醇(例如像乙二醇)与新戊二醇和/或2-丁基-2-乙基-1,3-丙二醇的摩尔比率是低于20%、更优选低于15%、甚至更优选低于12%、并且非常优选低于10%。具有过高水平的直链α,ω-醇的聚酯(例如像乙二醇)已被证明不太适合自发沉淀。在另外的实施方案中,粉末涂料粘合剂(特别是聚酯粉末涂料粘合剂)的沉淀可以完全出人意料地在充当沉淀控制剂的添加剂的存在下发生。这些添加剂的作用是在效应颜料表面上更有效地沉淀粉末涂料粘合剂。这表现在具有粘合剂的颜料的较高占有率和相应降低的二次沉淀物中。这些添加剂优选是基于马来酸酐与乙烯基芳族化合物、优选与取代或未取代的苯乙烯的梳形共聚物。此外,这些添加剂优选具有聚醚单元(eo和/或po),其通过酯、酰胺或酰亚胺基团连接到酸基团上。这些添加剂优选具有4至45mgkoh/g的酸值,更优选为5至40mgkoh/g。进一步优选地,它们是盐,其中酸值约等于胺值。这些种类的添加剂描述于例如ep2125909b1、ep2106412b1、ep2125910b1、ep2240543b1、de102010049642a1、ep2723779b1或ep2864373b1中。可商购的添加剂的实例是byk190、disperbyk2060、disperbyk2010、bykes80或byk187(均来自byk添加剂与仪器公司(bykadditives&instruments),abelstrasse26,d-46483韦塞尔,德国)。这些种类的添加剂通常主要用作润湿剂和分散剂,特别是在颜料糊剂的形成方面。在聚合物自发沉淀到如本发明中描述的效应颜料上期间其作为助剂的功能是不可预见的。在本发明的另一个实施方案中,在步骤a2)之后并且在加入粘合剂之前、并且任选地在加入添加剂之前,用粘合促进剂处理效应颜料。粘合促进剂、特别是官能硅烷,如以上更早地描述的。本发明的另一主题是本发明的效应颜料的用途。根据本发明的涂覆的效应颜料用于漆料(paints)、粉末涂料、印刷油墨、调色剂或塑料中。特别优选的是它们在粉末涂料中的用途。由于根据本发明使用的粘合剂是适合作为粉末涂料粘合剂的粘合剂,因此本发明的效应颜料可以被加工成粉末涂料中特别好的效果。在此,其中掺入效应颜料的粉末涂料的粘合剂可以与构成涂料的粘合剂相同或不同。在优选的实施方案中,可以选择聚酯树脂既作为效应颜料的涂料粘合剂,也作为粉末涂料的粘合剂。附图说明:图1:dsc图以5°k/min在从30℃至210℃的温度范围内以对温度的表示进行记录。该图仅显示朝向更高温度的线,并且没有反向的线。______(实线):预试验实例1:在从丙酮中沉淀后的粉末涂料cc4540-0,_._._.(点划线):预试验对比实例1:由制造商获得的粉末涂料cc4540-0,_.._.._.(双点划线):预试验实例1b:沉淀后的粉末涂料cc4540-0,第一次扫描后直接进行第二次扫描。图2:用于13cmasnmr概况测量(图2a)和13cnmrmas弛豫测量(图2b)的脉冲序列的示意图。图3:对于依照预试验对比实例1的无定形相(顶部)和依照预试验实例1的半结晶相(中间,虚线)以及实例1的涂覆的铝颗粒(底部,虚线)的预试验实例1(cc4540-0)的pxrd。测量在德拜-谢勒几何的stoestadip衍射仪上使用cukα1辐射(ge(111)单色器)进行。使用具有0.3mm直径的sio2毛细管。为了更好地概述,无定形样品和半结晶样品的测量数据向上移动了50000和25000计数。实例1的非常尖且高的峰可归因于铝基材的反射。图4:依照预试验对比实例1(底部,标记为“agm”;νrot=12.5khz)的无定形相、依照预试验实例1的半结晶相(中部,标记为“nd10%”;νrot=12.5khz)、以及依照实例1的涂覆的铝颗粒(顶部,标记为“50/50alu颜料”;νrot=8khz)的粘合剂cc4540-0的13ccpmas光谱。旋转边带用星号标记,并且各向同性化学位移归属于聚酯的酸和醇组分的特征化学结构单元。图5:弛豫程度相对于来自预试验实例1的半结晶聚合物的亚甲基的13c共振的等待时间的曲线图(粘合剂cc4540-0)。无定形部分和结晶部分的两个不同时间常数反映在具有显著不同斜率的两个区域中。图6:依照预试验对比实例10(底部,标记为“agm”;νrot=6khz)的无定形相以及依照预试验实例1的半结晶相(中部,标记为“nd”;νrot=6khz)的粘合剂cc2506的13ccpmas光谱。旋转边带用星号标记,并且各向同性化学位移归属于特征化学结构单元。图7:弛豫程度相对于来自预试验实例10的半结晶聚合物的亚甲基的13c共振的等待时间的曲线图(粘合剂cc2506-1)。无定形部分和结晶部分的两个不同时间常数反映在具有显著不同斜率的两个区域中。具体实施方式方面本发明进一步涉及包含根据权利要求或方面之一的涂覆的效应颜料的主题。根据方面1,本发明涉及一种涂覆的效应颜料,该效应颜料包含片状基材和施加在其上并且包含用于粉末涂覆材料的粘合剂的涂料;其特征在于,该粘合剂具有借助于13cnmrmas弛豫测量所确定的结晶和无定形部分,13c核的弛豫根据下式拟合为双指数弛豫其中结晶度c是在40%与85%之间的范围内,并且其中存在短平均弛豫时间t1s和长平均弛豫时间t1l,并且其中t1l是在从65至130s的范围内。根据本发明的方面2,根据方面1的涂覆的效应颜料优选包含具有45%至75%的结晶度c的粘合剂。根据本发明的方面3,在根据方面1和2中任一项的涂覆的效应颜料中,长平均弛豫时间t1l优选是在从70至110s的范围内。根据本发明的方面4,在方面1至3的涂覆效应颜料中,粘合剂优选不是lcst或ucst聚合物。根据本发明的方面5,在根据前述方面中任一项的涂覆的效应颜料中,使用cukα1作为x射线源并且使用具有0.5mm的狭缝宽度的锗(111)作为单色器,在德拜-谢勒几何(毛细管直径0.3mm)的pxrd衍射图中的粘合剂优选具有在从0.7°至2.0°(以2θ计)范围内的半峰全宽的结构化峰。根据本发明的方面6,在根据前述方面的涂覆的效应颜料中在5℃/min前进速率下在dsc图中优选具有至少一个吸热峰,其具有来自tmax=100℃至150℃的范围的最大值,并且还具有与该峰值相关来自从15j/g至80j/g范围的焓δh,该焓是根据粘合剂的量计算的。根据本发明的方面7,根据前述方面的涂覆的效应颜料优选通过根据方面19至33的方法生产。根据本发明的方面8,在根据前述方面中任一项的涂覆的效应颜料中,该粘合剂优选具有聚酯官能团并且通过熔融聚合制备。根据本发明的方面9,在根据方面8的涂覆效应颜料中,该粘合剂优选是含有酸和醇组分的聚酯,并且作为酸组分,主要部分取自由间苯二甲酸和对苯二甲酸及其混合物组成的组的单体。根据本发明的方面10,在根据方面8或9的涂覆的效应颜料中,基于所有的酸组分,该粘合剂优选包含至少14mol%的间苯二甲酸作为酸组分。根据本发明的方面11,在根据方面8至10中任一项的涂覆的效应颜料中,该粘合剂优选包含基于所有醇组分的分数为50至100mol%的新戊二醇和/或2-丁基-2-乙基-1,3-丙二醇作为醇组分。根据本发明的方面12,在根据方面11的涂覆的效应颜料中,关于聚酯的醇组分,该聚酯优选具有小于20%的乙二醇和/或二乙二醇与新戊二醇和/或2-丁基-2-乙基-1,3-丙二醇的摩尔比。根据本发明的方面13,在根据方面6至12中任一项的涂覆的效应颜料中,该粘合剂优选具有在从2至10的范围内的吸热熔化焓δh/δhagm的比率,其可以通过以5℃/min的前进速率进行dsc测量来确定,并且其基于相对于沉淀前(δhagm)的粘合剂的沉淀后(δh)的粘合剂的量。根据本发明的方面14,在根据方面6至13中任一项的涂覆的效应颜料中,该粘合剂优选表现出与未结晶状态相比的来自结晶状态的来自从40℃至75℃范围的dsc中的δtmax的增加。根据本发明的方面15,在根据前述方面中任一项的涂覆的效应颜料中,该片状基材优选取自由金属效应颜料、珠光颜料和干涉颜料、及其混合物组成的组。根据本发明的方面16,在根据前述方面中任一项的涂覆的效应颜料中,该片状基材优选为珠光颜料或者是涂覆有金属氧化物的金属效应颜料。根据本发明的方面17,在根据方面16的涂覆的效应颜料中,该金属氧化物优选取自由(二)氧化硅、氧化硅水合物、氢氧化硅、氧化铝、氧化铝水合物、氢氧化铝、及其混合物组成的组。根据本发明的方面18,在根据方面16和17中任一项的涂覆的效应颜料中,该片状基材优选是sio2涂覆的铝效应颜料。根据方面19,本发明涉及一种用于生产涂覆的效应颜料的方法,该涂覆的效应颜料包含片状基材和施用在其上的用于粉末涂覆材料的涂料,该方法包括以下步骤:a1)在温度tsol下在tsol的第一时间间隔内将可自发沉淀的粘合剂溶解在有机溶剂或溶剂混合物中,b1)随后将效应颜料加入来自a1)的溶剂或溶剂混合物中,同时分散该效应颜料,c1)在温度tinsol下在第二时间间隔tinsol内用该粘合剂涂覆该效应颜料,或者a2)将效应颜料分散在溶剂或溶剂混合物中,b2)随后向具有温度tsol的溶剂或溶剂混合物中加入可自发沉淀的粘合剂,c2)在温度tinsol下在时间间隔tinsol内用该粘合剂涂覆该效应颜料,d)从该溶剂或溶剂混合物中去除该涂覆的效应颜料,并且e)任选地干燥该涂覆的效应颜料。根据本发明的方面20,在根据方面22的方法中,优选地,特征在于,差值δt=tinsol–tsol是在从0℃至5℃的范围内并且存在自发沉淀。根据本发明的方面21,在根据方面19或20的方法中,优选地,特征在于,步骤c1)或c2)的温度tinsol是在从比该溶剂或溶剂混合物的沸腾温度低10℃至5℃的范围内。根据本发明的方面22,在根据方面21的方法中,优选地,特征在于,步骤c1)或c2)的温度tinsol是在从18℃至30℃的范围内。根据本发明的方面23,在根据方面219至22的方法中,优选地,特征在于,步骤a1)或b2)的温度tsol是在从比该溶剂或溶剂混合物的沸腾温度低10℃至5℃的范围内。根据本发明的方面24,在根据方面23的方法中,优选地,特征在于,步骤a1)或b2)的温度tsol是在从18℃至30℃的范围内。根据本发明的方面25,在根据方面19至24中任一项的方法中,优选地,特征在于,步骤c1)或c2)中的tinsol是在从0.5h至12h的范围内。根据本发明的方面26,在根据方面19至25中任一项的方法中,优选地,特征在于,该有机溶剂选自由丙酮、甲基乙基酮、乙酸乙酯、以及其混合物组成的组。根据本发明的方面27,在根据方面19至26中任一项的方法中,优选地,特征在于,该粘合剂不是lcst或ucst聚合物。根据本发明的方面28,在根据方面19至27中任一项的方法中,优选地,特征在于,该粘合剂具有聚酯官能团并且通过熔融聚合制备。根据本发明的方面29,在根据方面28的方法中,优选地,特征在于,该粘合剂是含有酸和醇组分的聚酯,并且作为酸组分,主要部分取自由间苯二甲酸和对苯二甲酸及其混合物组成的组的单体。根据本发明的方面30,在根据方面28或29的方法中,优选地,特征在于,基于所有的酸组分,该粘合剂优选包含至少14mol%的间苯二甲酸作为酸组分。根据本发明的方面31,在根据方面28至30中任一项的用于生产涂覆的效应颜料的方法中,优选地,特征在于,该粘合剂包含基于所有醇组分的分数为50至100mol%的新戊二醇和/或2-丁基-2-乙基-1,3-丙二醇作为醇组分。根据本发明的方面32,在根据方面31的用于生产涂覆的效应颜料的方法中,优选地,特征在于,关于聚酯的醇组分,其具有小于20%的乙二醇和/或二乙二醇与新戊二醇和/或2-丁基-2-乙基-1,3-丙二醇的摩尔比。根据本发明的另一方面33,根据方面19至32中任一项,步骤a2)之后,加入添加剂作为沉淀助剂,该添加剂优选为基于马来酸酐和含乙烯基的芳族化合物、优选苯乙烯的梳形聚合物。根据本发明的另一方面33,根据方面19至33中任一项,在步骤a2)之后并且在加入粘合剂之前、并且任选地在加入添加剂之前,用粘合促进剂处理效应颜料。根据本发明的方面34,将根据方面1至18的涂覆的效应颜料用于漆料、粉末涂料、印刷油墨、调色剂或塑料中。根据另一方面35,本发明涉及一种涂覆的效应颜料,该效应颜料包含片状基材和施加在其上的并且包含用于粉末涂覆材料的粘合剂的涂料;其特征在于,在5℃/min前进速率下的dsc图中,优选具有至少一个吸热峰,其具有来自tmax=100℃至150℃的范围的最大值,并且还具有与该峰值相关来自从15j/g至80j/g范围的焓δh,该焓是根据该粘合剂的量计算的。根据本发明的方面36,在根据方面35的涂覆的效应颜料中,该粘合剂优选具有在从2至10的范围内的吸热熔化焓δh/δhagm的比率,其可以通过以5℃/min的前进速率进行dsc测量来确定,并且其基于相对于沉淀前(δhagm)的粘合剂的沉淀后(δh)的粘合剂的量。根据本发明的方面37,在根据方面35或36中任一项的涂覆的效应颜料中,该粘合剂优选表现出与未结晶状态相比的来自结晶态的来自从40℃至75℃范围的dsc中的δtmax增加。根据本发明的方面38,在方面35至37的涂覆的效应颜料中,粘合剂优选不是lcst或ucst聚合物。根据本发明的方面39,在根据前述方面37至38中任一项的涂覆的效应颜料中,在使用cukα1作为x射线源并且使用具有0.5mm的狭缝宽度的锗(111)作为单色器的德拜-谢勒几何(毛细管直径0.3mm)的pxrd衍射图中的粘合剂优选具有在从0.7°至2.0°(以2θ计)范围内的半峰全宽的结构化峰。根据本发明的方面40,在根据前述方面35至39的涂覆的效应颜料中,该粘合剂优选具有借助于13cnmrmas弛豫测量来确定的结晶和无定形部分,13c核的弛豫根据下式拟合为双指数弛豫其中结晶度c是在40%与85%之间的范围内,并且其中存在短平均弛豫时间t1s和长平均弛豫时间t1l,并且其中t1l是在从65至130s的范围内。根据本发明的方面41,根据方面40的涂覆的效应颜料优选包含具有45%至75%的结晶度c的粘合剂。根据本发明的方面42,在根据方面40和41中任一项的涂覆的效应颜料中,长平均弛豫时间t1l优选是在从70至110s的范围内。根据本发明的方面43,根据前述方面34至42的涂覆的效应颜料优选通过根据方面19至33的方法生产。根据本发明的方面44,在根据前述方面34至43中任一项的涂覆的效应颜料中,该粘合剂优选具有聚酯官能团并且通过熔融聚合制备。根据本发明的方面45,在根据方面44的涂覆的效应颜料中,该粘合剂优选是含有酸和醇组分的聚酯,并且作为酸组分,主要部分取自由间苯二甲酸和对苯二甲酸及其混合物组成的组的单体。根据本发明的方面46,在根据方面44或45的涂覆的效应颜料中,基于所有的酸组分,该粘合剂优选包含至少14mol%的间苯二甲酸作为酸组分。根据本发明的方面47,在根据方面44至46中任一项的涂覆的效应颜料中,该粘合剂优选包含基于所有醇组分的分数为50至100mol%的新戊二醇和/或2-丁基-2-乙基-1,3-丙二醇作为醇组分。根据本发明的方面48,在根据方面47的涂覆的效应颜料中,关于聚酯的醇组分,该聚酯优选具有小于20%的乙二醇和/或二乙二醇与新戊二醇和/或2-丁基-2-乙基-1,3-丙二醇的摩尔比。根据本发明的方面49,在根据前述方面34至48中任一项的涂覆的效应颜料中,该片状基材优选取自由金属效应颜料、珠光颜料和干涉颜料、及其混合物组成的组。根据本发明的方面50,在根据方面49的涂覆的效应颜料中,该片状基材优选为珠光颜料或者是涂覆有金属氧化物的金属效应颜料。根据本发明的方面51,在根据方面50的涂覆的效应颜料中,该金属氧化物优选取自由(二)氧化硅、氧化硅水合物、氢氧化硅、氧化铝、氧化铝水合物、氢氧化铝、及其混合物组成的组。根据本发明的方面52,在根据方面50和16中任一项的涂覆的效应颜料中,该片状基材优选是sio2涂覆的铝效应颜料。根据本发明的方面53,将根据方面34至52之一的涂覆的效应颜料用于漆料、粉末涂料、印刷油墨、调色剂或塑料中。本发明的另一主题是根据以下方面的聚合物。根据本发明的方面54,优选是聚合物,其可以是粘合剂并具有聚酯官能团并且在溶剂或溶剂混合物中自发沉淀。根据本发明的方面55,根据方面54的聚合物优选是用于粉末涂料的粘合剂。根据本发明的方面56,根据方面54和55中任一项的聚合物的优选地特征在于,该粘合剂是含有酸和醇组分的聚酯,并且作为酸组分,主要部分取自由间苯二甲酸和对苯二甲酸及其混合物组成的组的单体。根据本发明的方面57,根据方面54至56中任一项的聚合物优选特征在于,基于所有的酸组分,该粘合剂包含至少14mol%的间苯二甲酸作为酸组分。根据本发明的方面58,根据方面54至57中任一项的聚合物优选特征在于,该粘合剂包含基于所有醇组分的分数为50至100mol%的新戊二醇和/或2-丁基-2-乙基-1,3-丙二醇作为醇组分。根据本发明的方面59,根据方面58的聚合物优选特征在于,关于聚酯的醇组分,该聚酯具有小于20%的乙二醇和/或二乙二醇与新戊二醇和/或2-丁基-2-乙基-1,3-丙二醇的摩尔比。根据本发明的方面60,根据方面54至59中任一项的聚合物优选具有借助于13cnmrmas弛豫测量来确定的结晶和无定形部分,13c核的弛豫根据下式拟合为双指数弛豫其中结晶度c是在40%与85%之间的范围内,并且其中存在短平均弛豫时间t1s和长平均弛豫时间t1l,并且其中t1l是在从65至130s的范围内。根据本发明的方面61,根据方面60的聚合物优选具有45%至75%的结晶度c。根据本发明的方面62,根据方面60和61中任一项的聚合物,长平均弛豫时间t1l优选是在从70至110s的范围内。根据本发明的方面63,根据方面54至62的聚合物粘合剂优选不是lcst或ucst聚合物。根据本发明的方面64,根据前述方面54至63中任一项的聚合物优选是在使用cukα1作为x射线源并且使用具有0.5mm的狭缝宽度的锗(111)作为单色器的德拜-谢勒几何(毛细管直径0.3mm)的pxrd衍射图中优选具有在从0.7°至2.0°(以2θ计)范围内的半峰全宽的结构化峰的聚合物。根据本发明的方面65,根据前述方面54至64的聚合物在5℃/min前进速率下在dsc图中优选具有至少一个吸热峰,其具有来自tmax=100℃至150℃的范围的最大值,并且还具有与该峰值相关的来自从15j/g至80j/g范围的焓δh,该焓是根据粘合剂的量计算的。根据本发明的方面66,该聚合物优选通过根据方面69至81中任一项的方法制备。根据本发明的方面67,根据方面73至85中任一项的聚合物,该有机溶剂优选选自由丙酮、甲基乙基酮、乙酸乙酯、以及其混合物组成的组。根据本发明的方面68,根据方面54至67中任一项的聚合物优选用于涂覆片状效应颜料。根据方面69,本发明涉及一种用于生产聚合物的方法,该方法包括以下步骤:c1)在温度tsol下在tsol的第一时间间隔内将可自发沉淀的粘合剂溶解在有机溶剂或溶剂混合物中,c2)随后在温度tinsol下在第二时间间隔tinsol内沉淀该聚合物,其中差值δt=tinsol–tsol是在从0℃至5℃的范围内并且存在自发沉淀,d1)从该溶剂或溶剂混合物中去除该聚合物,并且e)任选地干燥该聚合物。根据本发明的方面70,在根据方面69的方法中,优选地,特征在于,步骤c1)或c2)的温度tinsol是在从比该溶剂或溶剂混合物的沸腾温度低10℃至5℃的范围内。根据本发明的方面71,在根据方面70的方法中,优选地,特征在于,步骤c2)的温度tinsol是在从18℃至30℃的范围内。根据本发明的方面72,在根据方面69至71的方法中,优选地,特征在于,步骤c1)的温度tsol优选是在从比该溶剂或溶剂混合物的沸腾温度低10℃至5℃的范围内。根据本发明的方面73,在根据方面72的方法中,优选地,特征在于,步骤c1)的温度tsol是在从18℃至30℃的范围内。根据本发明的方面74,在根据方面69至73中任一项的方法中,优选地,特征在于,步骤c2)中的tinsol是在从0.5h至12h的范围内。根据本发明的方面75,在根据方面69至74中任一项的方法中,优选地,特征在于,该有机溶剂选自由丙酮、甲基乙基酮、乙酸乙酯、以及其混合物组成的组。根据本发明的方面76,在根据方面69至74中任一项的方法中,优选地,特征在于,该粘合剂不是lcst或ucst聚合物。根据本发明的方面77,在根据方面69至76中任一项的方法中,优选地,特征在于,该粘合剂具有聚酯官能团并且通过熔融聚合制备。根据本发明的方面78,在根据方面77的方法中,优选地,特征在于,该粘合剂是含有酸和醇组分的聚酯,并且作为酸组分,主要部分取自由间苯二甲酸和对苯二甲酸及其混合物组成的组的单体。根据本发明的方面79,在根据方面77或78的方法中,优选地,特征在于,基于所有的酸组分,该粘合剂优选包含至少14mol%的间苯二甲酸作为酸组分。根据本发明的方面80,在根据方面77至79中任一项的用于生产涂覆的效应颜料的方法中,优选地,特征在于,该粘合剂包含基于所有醇组分的分数为50至100mol%的新戊二醇和/或2-丁基-2-乙基-1,3-丙二醇作为醇组分。根据本发明的方面81,在根据方面80的用于生产涂覆的效应颜料的方法中,优选地,特征在于,关于聚酯的醇组分,其具有小于20%的乙二醇和/或二乙二醇与新戊二醇和/或2-丁基-2-乙基-1,3-丙二醇的摩尔比。实例a1预试验1至15:在预试验中,在室温下将各种不同的聚酯粘合剂(商业上作为粉末涂料粘合剂提供)溶解在溶剂如丙酮、甲基乙基酮(mek)或乙酸乙酯中,并且然后研究可能的自发沉淀。为此目的,将该溶剂装入玻璃烧杯中,如果需要,调节温度,并且在搅拌下加入粘合剂粉末(典型地约为溶剂量的1/6)。所有表现出自发沉淀的粘合剂首先全部溶解而没有残余物,但在这种情况下采取略带灰色的溶液形式,表明至少部分胶体状态。不显示自发沉淀的粘合剂(对比实例)总是溶解在无色透明溶液中。自发沉淀发生在不同时间:这些时间可能总计为几分钟到约48小时。通过vwr滤纸454(颗粒截留率:12-15μm)分离沉淀的聚合物,并且在真空干燥箱中在50℃下干燥6h。然后可以进一步分析这些聚合物粉末。对许多所选择的聚合物的系统研究表明,完成的沉淀为至少90%,并且在大多数情况下大部分是定量的。在这点上,所有结果总结在表1中。预试验对比实例1至3:将粘合剂cc340、cc2818-0(均为allnex)和uralacp1580(dsm)溶解在丙酮、乙酸乙酯或甲基乙基酮中,但不再沉淀。因此,在本发明的意义上,这些粘合剂不是可自发沉淀的。预试验对比实例4至6:在预试验中,试图在室温以及在直到低于相应的沸腾温度5℃的各种温度下将半结晶粉末涂料聚酯树脂uvcoat9010、additolp791和crylcoat-8078-0(均来自allnex)溶解在乙酸乙酯、丙酮和甲基乙基酮中。然而,树脂不能溶解到与本发明相应的足够程度。因此,在本发明的意义上,这些粘合剂不是可自发沉淀的。对这些树脂研究其单体组成(参见表1)并且由dsc表征(见表2),并且可以进行分析。a2粘合剂的结构这些粘合剂通过以下聚合物分析的常规方法分析其单体组成,例如像gc-质谱、ir光谱和nmr(1h、13c、以及2-dnmr)。在这些分析中,以非常少量存在的一些酸单体组分被共同归入“其他酸”。表1提供了所研究粘合剂的概述。在此还非常一般地报道了沉淀时间以及单体组分分析的结果。对于在此的每种样品,间苯二甲酸或对苯二甲酸的量(预试验对比实例3)在每种情况下取100份,并且所有其他组分与它们成比例地表示。在另外的预试验实例中,使用适用于粉末涂料的各种不同聚酯树脂重复实例1的程序。在室温下在丙酮中的溶解试验结果以及分析测定的单体组成在表1中示出。在溶剂如乙酸乙酯或甲基乙基酮(mek)中也获得了类似的结果。在非常非极性的溶剂如氯仿中,这些粘合剂完全溶解,得到澄清溶液。然而,没有沉淀。总而言之,在所研究的二十一种聚酯粘合剂中,十五种显示出自发沉淀的行为(预试验实例1-15),而三种粘合剂虽然溶解在溶剂中,但不再经历自发沉淀(预试验对比实例1-3),并且另外三种粘合剂不是可溶的(预试验对比实例4-6)。这些粘合剂中的大多数也在乙酸乙酯和甲基乙基酮中进行试验,得到类似的结果。与新戊二醇相比,预试验对比实例1-3的粘合剂显示出高分数的乙二醇和/或三羟甲基丙烷。由其制造商作为半结晶粉末涂料粘合剂销售的另外三种粘合剂不能充分溶解(预试验对比实例4-6)。这些粘合剂具有己二酸(预试验实例5)或1,12-十二烷酸作为其酸组分的主要部分。存在的醇组分(在可分析时)不包括可检测量的新戊二醇,而是乙二醇(预试验实例4)或顺式-和反式-二羟甲基环己烷的混合物。显然,这些粘合剂已经具有结晶部分,使得它们不能溶解,并且因此也不再表现出任何“自发聚合”。对预试验粘合剂的dsc研究:对于预试验的本发明的实例,另外进行dsc研究。在这些研究中,将沉淀并且干燥的粘合剂(预试验实例)以及从制造商获得的用于对比的未沉淀干燥粘合剂(预试验对比实例)的小的确定量(几mg)都进行称重并且进行dsc测量(仪器制造商:德国耐驰公司(netzsch);型号sta449f3jupiter;对比标准:铟)。在此的前进速率是5°k/min,并且温度范围通常从30℃延伸至250℃。没有等待时间的样品随后以5°k/min冷却至40℃,并且在该温度下20分钟的等待时间后,以5°k/min记录第二次温度运行直至250℃。测量在空气下进行,因为不能确定没有与氧气的反应性。在这些研究中,在未沉淀的对比样品的情况下,在每种情况下存在具有在超过60℃至约75℃范围内的最大值并且具有约55℃至低于70℃的相应tonset的吸热峰。作为一般规则,位于这些峰下方的是玻璃化转变温度tg,其在dsc中已知在基线的偏移中表现出来。该玻璃化转变温度可以主要归属为聚合物的无定形部分。dsc中的这种行为对于这些聚酯粘合剂是已知的并且完全是常规的。该峰可能归因于某些小晶体部分的熔化。相比之下,在自发沉淀的样品的情况下,发现了显著更大的吸热峰,其具有在从约120℃至约135℃的范围内的最大tmax并且具有约100℃至124℃的相应tonset。在此同样,也可能存在低于峰值的玻璃化转变温度。所有样品的反向温度运行总是基本上没有结构,没有明显可识别的峰。这清楚地表明,作为自发沉淀的结果,强烈移动和增大的吸热峰可归因于所研究的聚酯树脂的结晶部分的显著增加。当对自发沉淀的样品进行第一次温度循环后的第二次测量时,发现的行为总是与未沉淀的样品相似。作为举例,图1显示出预试验实例1的正向温度运行。由制造商得到的未沉淀聚合物(预试验对比实例1)显示出具有在70.1℃处的最大值的吸热峰。相比之下,沉淀的聚合物表现出在126.2℃下的吸热峰和高得多的焓(预试验实例1)。如果再次记录该聚合物的dsc光谱,则在向后运行至30℃后,随后的dsc扫描与未沉淀的聚合物(预试验对比实例1b,在71.5℃下的tmax)的dsc扫描高度相当。作为自发沉淀的结果而获得的晶体结构显然在较高温度下被不可逆地破坏。然而,如果将该聚合物再溶解在溶剂中并且自发沉淀,那么将再次得到类似于预试验实例1的dsc曲线(图1中未示出)。在这种意义上,结晶程序是可逆的。对于所有dsc试验,tga测量同时进行。对于一些样品,发现质量减少在从1至约2wt%的范围内。在此通常有可能在约50℃的温度下观察第一步骤。该步骤可以解释为此时从沉淀的聚合物中除去残留的痕量溶剂。积分峰值以计算相关的焓δh。表2给出了所获得的测量数据的概述。它显示了起始温度tonset、最大温度tmax以及与未沉淀的粘合剂样品(预试验对比实例)和沉淀样品(预试验实例)的吸热峰的峰(在减去常规背景校正之后)相关的焓δh。还示出了这些参数的变化,在每种情况下,从沉淀的粘合剂的值中减去未沉淀的粘合剂的值。在焓的情况下,由沉淀的与未沉淀的聚酯树脂形成比率。作为自发沉淀的结果,tmax值的这种大的温度变化、以及吸热峰的相关焓的急剧增加可以归因于聚合物的结晶部分的显著增加。表2:对于来自沉淀粘合剂(预试验实例)和未沉淀粘合剂(预试验对比实例)的dsc测量的tonset、tmax和δh参数从120℃至超过132℃的熔化范围的非常高的值在文献中类似地仅被称为所谓的“半结晶”聚酯(ep0521992b1,us4,217,426)。在那里,还报告了类似的焓值。然而,在此显著的是,基于单体组合物,粘合剂更多地称为“无定形”粘合剂(例如,us6,660,398b1)。通过自发沉淀实现的结晶度似乎非常高。在105.1和191.1j/g的预试验对比实例4和5(仅在第一次扫描中测量)的焓值仍然高于沉淀后本发明实例的所有测量的焓值。这似乎反映了更高程度的结晶。这些粘合剂显然是如此高度结晶的,以至于它们不能溶解于在此使用的溶剂中,并且因此不进入自发结晶。b效应颜料的涂覆:实例1:使用开口的1升夹套反应器,用低温恒温器冷却至15℃。将60.0g的具有35μm的平均粒度d50的sio2涂覆的铝效应颜料(pcs3500,由eckartgmbh可获得的)引入并且悬浮在250g的丙酮中。通过振动斜槽,在60min的时间内,将63.0g的名称为crylcoat4540-0的含羧基的饱和聚酯树脂(制造商:allnexs.à.r.l.)加入该悬浮液中。添加结束后,将悬浮液搅拌2小时,并且然后通过吸滤器排出。将得到的沉淀物在可加热的duplex捏合机(型号hkd-t06d,来自gmbh&co.kg)中在80毫巴和35℃下减压干燥1小时,并且然后通过具有小于100μm的筛孔尺寸的筛子进行筛分。实例2:重复实例1的程序,但使用具有名称siralespe7499的树脂作为聚酯树脂(制造商:sirindustrialespa)(对应于预试验14)。实例3:重复实例1的程序,但使用具有名称cc2506-1的树脂作为聚酯树脂(制造商:allnexs.à.r.l.)(对应于预试验10)。实例4:重复实例1的程序,但使用60.0g的sio2涂覆的金青铜颜料dorolanreichbleichgold17/0作为效应颜料。实例5:重复实例1的程序,但使用60.0g珠光颜料luxane221(来自德国eckartgmbh)作为效应颜料。实例6:重复实例1的程序,但使用乙酸乙酯代替丙酮作为溶剂,并且在加入粘合剂之前,向金属效应颜料悬浮液中加入1.0g的disperbyk2060,并且在加入粘合剂之前搅拌1h。添加结束后,将悬浮液搅拌2小时,并且此后将该悬浮液静置1h。形成仅略微混浊的溶剂上清液,同时涂覆的金属效应颜料经历沉降。如果在不加入添加剂的情况下用根据实例1的配方采用相应的程序,则上清液更加浑浊。在光学显微照片和sem显微照片中可以看出,在实例6的情况下,金属效应颜料与实例1的情况下相比用聚合物更好并且更均匀地涂覆,其中观察到一些二次沉淀。实例7:重复实例6的程序,但在将disperbyk2060加入金属效应颜料的悬浮液中之前,将其与2.0g的dynasylanglymo(来自赢创工业有限公司(evonikindustriesag))混合并且在室温下搅拌1h。聚合物沉淀物的量与实例6相当。对比实例1(基于ep1699884b1):将195g的聚酯树脂crylcoat2818-0(来自allnexs.à.r.l.)和作为固化组分的105g的具有名称vestagonbf1320(制造商:evonik)的多异氰酸酯加合物溶解于1800g的丙酮内,并且分散200g的sio2涂覆的铝效应颜料(pcs3500,从eckartgmbh可获得的)。在其温度为55℃的空气流中,以30g/min的速率以2.5巴的喷雾压力在喷雾干燥器中喷雾该分散体。干燥后,得到380g涂覆的效应颜料的产率。对比实例2:如对比实例1中涂覆,但使用200g珠光颜料luxane221(来自德国eckartgmbh)作为基材。对比实例3:使用基于de10243438a1的ucst聚合物如实例1中悬浮金属效应颜料,但使用250g的去离子水作为溶剂。所加入的聚合物是63g的vpc55k65w(来自巴斯夫公司(basf)的ucst聚合物,其具有约65℃的在水中的浊点温度)。然后在1小时的过程中将温度升至75℃,并将混合物在该温度下放置1小时。随后,在仍然热的同时过滤分散体。与本发明实例相反,所得沉淀物非常坚韧,可归因于颜料颗粒的显著附聚。附聚物被证明是如此大量的,以至于颜料的进一步测试显得不是有用的。在另外的对比试验中,改变沉淀温度,并且使用乙酸乙酯代替水作为溶剂;然而,总是产生附聚物。b1性能特性b1.1储存稳定性试验:在基于dineniso8130-8的方法中,在每种情况下将50g来自本发明实例和对比实例的颜料粉末引入可密封的铝罐中,并在60℃的加热箱中储存28天。在该时间结束后,相对于在室温下储存的对比样品,评估稠度(consistency)并根据以下标准进行评分。表3:储存试验是基于以下评估体系:表4:储存试验结果:样品储存试验指数实例10实例20实例30实例40实例50实例60实例70对比实例13对比实例23所有本发明实例以及对比实例2至5均显示出优异的储存稳定性(指数0)。然而,对比实例1和2的储存稳定性仅评价为指数3。改进的储存稳定性很可能归因于本发明实例的粘合剂涂料的结晶度增加。b1.2:聚合物涂料的粘附性的坚固性:本发明产品的一个重要标准是树脂涂料在效应颜料表面上的粘附性。树脂涂料在干燥和筛分的生产步骤期间以及在本发明产品的使用者的进一步加工步骤中经受严重暴露。为了确定效应颜料表面上的树脂涂料的粘附性,使本发明实例1、6和7的样品经受颗粒数和粒度的测定。在这种情况下,尺寸测量在sysmexfpia-3000s中进行。ce50是其中累积尺寸分布中测量颗粒的50%等效圆尺寸小于该值的尺寸值(数均)。该仪器具有用于分散样品的超声单元。这种分散在可调节幅度下并且在30khz的频率下合计为15瓦。在没有超声暴露的情况下测量样品,并且为了模拟处理步骤期间的机械负载,用超声以20%和60%幅度进行测量。下表5阐明了以下结果:表5:ce50值和所测量的颗粒数(使用sysmexfpia3000s测定):实例6的样品始终显示出比实例7的那些更低的ce50值和更高的颗粒数。这可归因于聚合物涂料的二次沉淀增加。该二次沉淀通常显著小于效应颜料。随着幅度增加并因此增加超声处理中的能量输入,ce50值在两种情况下均下降。在此,显然,颜料尺寸减小和/或存在聚合物涂料的部分磨损。因此,在实例7的情况下聚合物涂料在颜料表面上比在实例6的情况下更好地粘接,这可能归因于环氧硅烷的粘附的介导。b2分析调查b2.1dsc测量:类似于预试验,使用来自耐驰公司(netzsch,型号sta449f3jupiter)的仪器对本发明实例进行dsc研究。分析涂覆的效应颜料的粘合剂含量。dsc测量中的曲线轮廓对应于预试验的曲线轮廓。吸热峰中的起始温度和最高温度的位置在很大程度上对应于相应的预试验的位置。对于这些量的粘合剂,转化吸热峰的焓。用预试验发现了非常好的匹配,具有在+/-5%范围内的偏差。b2.2pxrd粉末衍射图所有粉末衍射测量均在stoestadip衍射仪上以德拜-谢勒几何在透射中室温下进行。该衍射仪配备有铜辐射和锗单色器(111),因此只有cukα1带用于衍射。源功率被设定为40kv和40ma,以确保背景与布拉格散射的最佳比率。为了限制光束发散,在样品前面使用0.5mm的准直器。所有测量均在5°与70°之间的2θ范围内使用具有64x8mm的检测窗口的mythen检测器(来自dectris)进行,其对应于以2θ计的19°开口角。检测器以0.2°步进行操作,每个步骤的曝光时间为90s。对各个测量值进行求和。这导致每个样品的8h和8min的测量时间。这些样品在0.3mm硼硅酸盐玻璃毛细管(来自hilgenberg,mark-tubes)中制备。b2.3c13masnmr概述测量在来自布鲁克公司(bruker)的avanceiiihd傅里叶变换nmr光谱仪上记录所有高分辨率一维13cnmr光谱(例如,图4和6)。该光谱仪配备有三个通道和具有9.4t的磁场强度的“宽口径”磁铁(低温磁铁)。这相当于1h核的400.13mhz拉莫尔频率以及13c核的100.62mhz拉莫尔频率。将样品置于4mm二氧化锆转子中,并且转移到来自bruker的4mmmas双共振样品头中。在测量期间,样品保持器的旋转频率对于纯聚合物(预试验实例1、10和15以及预试验对比实例1、10和15)为12.5khz,并且对于涂覆的铝颗粒为8.0khz(实例1)。使用13c通道上的cw脉冲和1h通道上的形状脉冲通过交叉极化实现激发。借助于形状脉冲,1h极化以常规方式转移到13c核。图2a概述了所使用的脉冲序列。在形状脉冲期间,在hartmann-hahn区段期间脉冲幅度线性增加了两倍。将起始质子-90°脉冲调节至2.5μs的长度,并且在hartmann-hahn区段期间1h和13c的章动频率为65khz(平均形状)和53khz。如已知的,用于将磁化从质子转移到13c核的hartman-hahn条件是如下:<ν(1h)>–ν(13c)=n*νrot,其中通常地n=±1。hartmann-hahn区段的3ms持续时间(接触时间)产生最佳转移效率。选择1s的重复时间,以便在所有情况下允许超过90%的质子磁化弛豫成热平衡。fid的记录伴随着借助于spinal64序列的宽频(broadband)质子去耦(描述于b.m.fung,a.k.khitrin,k.ermolaev,j.magn.reson.[磁共振杂志]2000,142,97-104),其中质子章动频率被调节到73khz,并且各个相移之间的时间间隔被调节至7.2μs。在40ms的采集时间内,fid(自由感应衰减)记录了4096个点。虽然对于纯聚合物样品(预试验)累积了4096个瞬变,但是在涂覆的铝颗粒(实例1)的情况下,低灵敏度需要51600次重复。通过随后的傅立叶变换以4096个点的“零填充”并使用具有10hz的半峰全宽的洛伦兹线进行折叠来获得1d光谱。相对于标准tms(0ppm)报告13c的所有化学位移。对预试验实例1、10和17(自发沉淀的聚合物)以及相应的预试验对比实例1、10和17(原始状态的聚合物样品)还有实例1的聚合物样品准备13cmasnmr概述测量。作为举例,图4示出了实例1、预试验实例1和预试验对比实例1的光谱。该图还包括聚酯结构元素的化学式及其在nmr谱中的峰的归属。作为举例,图6示出了实例10、预试验实例10和预试验对比实例10的光谱。与图4相比更清楚地,由于粘合剂的高对苯二甲酸分数,有可能在130ppm的范围内形成双结构。在每种情况下,沉淀的半结晶聚合物清楚地显示出比未沉淀的无定形聚合物更有效地结构化13cnmr光谱。在涂覆的铝颜料(实例1)的情况下,结构不像纯聚合物(预试实例1)情况下的完全过于明显。这可能是由于该样品中更大的敏感性差异。对于无定形样品(预试验对比实例1、10和17),13cmas光谱的半峰全宽是在250与350hz之间,并且对于半结晶聚合物(预试验实例1、10和17)是在100与180hz之间。涂覆的al颗粒(实例1)的半峰全宽是在180与270hz之间。由于不可能仅从半峰全宽来确定结晶度,因此也进行13cnmrmas弛豫测量。b2.413cnmrmas弛豫测量:13c自旋晶格弛豫(t1)的所有测量均在来自bruker的avanceii傅里叶变换nmr光谱仪上进行。此光谱仪配备有三个通道和具有7.05t的磁场强度的“宽口径”磁铁。这对应于1h和13c的300.15mhz和75.48mhz的拉莫尔频率。将样品引入7mm二氧化锆转子中,并且转移到来自bruker的7mmmas三共振样品头中。在测量期间,样品保持器的旋转频率为6khz。选择低旋转频率以便通过淬灭质子浴中的自旋扩散来最小化旋转频率对自旋晶格弛豫的影响。记录以饱和序列进行。对于预试验实例和对比实例的纯聚合物,饱和区段含有10个具有3.5μs长度的13c-90°脉冲,并且对于涂覆的al颗粒(实例1)具有7.0μs,并且还具有在其之间的20ms的失相间隔(dephasinginterval)。在饱和区段结束时,剩余磁化是小于0.1%,其中下降磁化的最大部分是由甲基共振的快速弛豫引起的。饱和区段之后是在100μs与1400s之间的可变等待时间,其中纵向磁化再次累积,这取决于该等待时间和时间常数t1。等待时间以指数差异选择,从而在每个完整的十个中测量六个值。为了检测重新建立的极化,后者借助于13c通道上的90°读取脉冲转换为横向可观察的磁化,同样,该通道具有3.5μs和7.0μs的长度。在用spinal64序列宽频质子去耦下,以15ms的采集时间和2048个点记录得到的fid。对于后者,质子章动频率为75khz,并且各个相移之间的时间间隔为7.4μs。在纯聚合物样品(预试验实例和对比实例)的情况下,在每个等待时间记录每个fid的64个瞬变。对于涂覆的al颗粒(实例1),每个fid的重复次数为1200。图2b给出了所使用脉冲序列的示意图。为了使得自旋晶格弛豫能够进行频率分辨评估,将获得的伪2d数据沿f2域进行傅里叶变换。为此目的,使用2048个点的“零填充”和具有洛伦兹线的折叠,其具有50hz的半峰全宽。再次相对于标准tms(0ppm)报告13c的所有化学位移。由此获得的光谱通过对于仍然不含有显著光谱强度的最短等待时间使用“三次样条(cubicspline)”插值调节光谱进行基线校正。然后将得到的拟合曲线从所有其他光谱中减去。这之后,通过强度的总和对特征化学结构单元的共振进行积分。使用来自mathworks的程序包matlab版本r2014b(8.4.0.150421)进行基线校正和积分。积分了以下光谱范围:17-28ppm(me-)、61-78ppm(-ch2-)、30-40ppm(cq)、124-140ppm(-caromatic-)、和160-170ppm(-co2-)。借助于等式(1)将积分强度1作为等待时间t的函数双指数地调节。这是使用levenberg-marquardt算法以其在程序gnuplot(版本4.6补丁级别5)中的实施完成,其中对于最小平方误差之和中的每个数据点,加权为1.0。平衡磁化m0分别作为最大等待时间的强度平均值进行调整,并且在整体精化(refinement)中不再改变。在所使用的levenberg-marquardt算法的情况下,仅改变变量a、c、t1s和t1l。为此目的,关于测量,至少对于最后三个测量值而言,必须牢记强度不再系统地上升。参数a考虑了非零初始磁化,并且因此可以被视为精化的技术变量,否则其与用于确定聚合物和涂覆的al颗粒中的结晶部分无关。参数a反映饱和区段和基线校正的效率,并且在t=0时完全消除磁化的时候应该接近1.0。对于在此进行的测量,在0.92与1.1之间变化。因此,为了确定结晶度,存在剩余的三个变量——自旋晶格弛豫的两个时间常数t1s和t1l、以及加权c。t1s和t1l差别非常显著,并且因此易于分开。与粉末衍射图(图3)一起,这表明样品具有不均匀性,其中可以区分无序(无定形)和有序(结晶)区域。基于更有效的弛豫,将具有时间常数t1s的快速自旋晶格弛豫分配给其中预期更大的物力(dynamism)的无定形区域。相应地,t1l是结晶区域的特征。相应地,常数c描述了原子百分比中结晶区域的分数。作为举例,图5和7示出了弛豫程度φ=(m0-m(t))/m0相对于等待时间t的典型曲线。y轴以对数方式绘制,并且因此各个弛豫分量显示为直线。所进行的驰豫测量的评估结果列于表6中。在这种情况下,甲基的信号被忽略,因为众所周知,由于c原子与相邻h原子之间的自旋旋转弛豫,它们非常快速地弛豫,这意味着在结晶与无定形部分之间没有检测到差异。表613cmas弛豫测量的评估结果为了评价,在每种情况下使用酸组分的芳族基团(简称car)的酸基团(简称-co2-)的、以及新戊醇组分的亚甲基(简称-ch2-)和季c原子(简称cq)的13c核的弛豫。如已知的,由于它们常见的非常快速的弛豫,所以没有使用甲基。对于所确定的值c、t1s和t1l,对应于该方法的通常程序,在每种情况下使用所有单独组的平均值。归属于无定形部分的短弛豫频率与归属于结晶部分的长弛豫频率之间存在明显差异。本发明实例1和预试验实例1、10和15表现出比相应的预试验对比实施方案(39.7至47.8s)高得多的弛豫时间t1l(73.6至98.25s)。然而,在参数c中,不存在像参数t1l情况中明显的清楚界定。研究的所有样品具有约70%的结晶度c。总的来说,这些发现可以解释为意指未沉淀的粘合剂聚合物已经具有非常低的空间范围的某种预序(preorder)。然而,在沉淀的粘合剂聚合物的情况下,有序区域在空间上急剧增加,导致更长的弛豫时间t1l。因为在此概述的13cnmr弛豫测量是在分子水平上极其敏感的方法,所以具有非常低阶的区域也有可能导致高c值。使用该方法,从少至约3至4个单位晶胞中检测聚合物中的局部有序。这些对nmr结果的解释与pxrd研究的结果非常吻合。在那里,预试验对比实例的未沉淀样品始终显示非结构化光谱。在此已经存在的聚合物的预序是明显的,虽然在空间方面不够明显给出德拜-谢勒几何中的信号。用基于更长距离相互作用的这种方法,仅当存在至少约20个结晶区域的单位晶胞时才获得结构化信号。在纯聚合物样品(预试验实例)和聚合物沉淀到al片(实例1)二者的情况下,沉淀的聚合物给出了清晰结构化的光谱。在nmr弛豫试验的情况下,沉淀聚合物的结晶部分的域尺寸急剧增加导致观察到更高的弛豫时间t1l。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1