一种有机荧光材料及其制备方法以及在检测神经性毒剂中的应用
1.本发明属于有机半导体纳米材料领域,具体涉及一种有机荧光材料及其制备方法以及在检测神经性毒剂中的应用。
背景技术:
2.化学战剂是一种具有剧烈毒性、能大规模地毒害或杀伤敌方人畜和植物的化学武器,曾经作为大规模杀伤性武器被多次使用。化学战剂主要分为神经性毒剂(nerve agents)和糜烂性毒剂(sulfur mustard)两大类。神经性毒剂是一类剧毒的有机磷酸酯或有机磷酸酯类化合物,也称为有机磷毒剂。神经性毒剂进入人体后作用于神经系统,通过抑制胆碱酯酶活性从而引起乙酰胆碱的蓄积,胆碱能使神经过度兴奋,最后导致呼吸、循环系统衰竭死亡。神经性毒剂根据化学结构及其特点可以分为两类:一类为g类毒剂,以呼吸道吸入为主要中毒途径,如沙林(sarin,gb)、梭曼(soman,gd)和塔崩(tabun,ga)等;另一类为v类毒剂,其毒性远超于g类毒剂。由于p-cl键的键能小于p-f,而真实的毒剂都是含有p-f键的,所以氯磷酸二乙酯(dcp)并不适用于作为神经性毒剂模拟物,实验室目前是用dcnp作为神经性毒剂模拟物。上述常见神经性毒剂及其模拟物结构如下所示:
3.神经性毒剂
4.模拟物
5.现有的文献报道的主要是针对神经性毒剂的检测方法,包括比色检测方法、表面声波法、酶化验法、干涉法、化学反应法等。然而上述方法存在反应缓慢、缺乏特异性、灵敏度低、操作复杂等缺点。采用荧光方法对化学战剂(cwas)进行检测,具有操作简便、响应灵敏、信号反应快、检测的特异性、制成小的荧光器件便于携带等优势。尽管如此,现有的荧光检测方法多数采用化学反应前后荧光光谱的变化来进行区分,这种情况下酸(例如盐酸(hcl))及其他含磷的有机物会对检测造成很大的影响。并且这种方法需要配成一定浓度的溶液,反应时间长,影响了检测的时效性,并且材料不能重复利用,经济效益不高,因此有待开发时效性和重复性好的荧光检测方法。dcp极易分解为hcl,因此dcp和hcl一样是两种重要的干扰物,经常会对荧光材料产生一种错误的响应信号。我们设计一种荧光检测材料,利用一种非共价键作用使其能够快速且有效的检测神经性毒剂并且可以排除酸及其它干扰物。
技术实现要素:
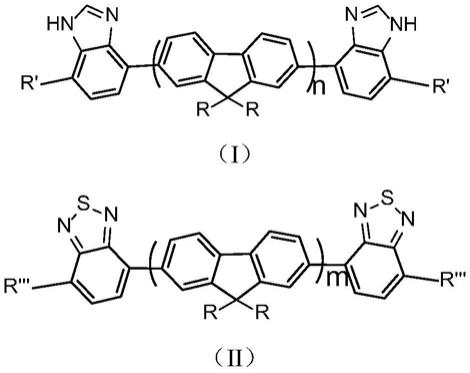
6.为解决上述问题,本发明提供一种有机荧光传感材料,其由如下式(i)所示化合物和式(ii)所示化合物通过共组装得到,
[0007][0008][0009]
其中,
[0010]
n选自3-5的整数;
[0011]
m选自3-5的整数;
[0012]
每个r相同或不同,彼此独立地选自无取代、或任选被一个、两个或多个ra取代的c
1-12
烷基、c
6-14
芳基、c
6-14
芳基-c
1-12
烷基;
[0013]
每个r'相同或不同,彼此独立地选自无取代、或任选被一个、两个或多个rb取代的c
6-14
芳基、c
6-14
芳基-c
1-12
烷基、5-14元杂芳基、5-14元杂芳基-c
1-12
烷基;
[0014]
每个r”'相同或不同,彼此独立地选自无取代、或任选被一个、两个或多个rc取代的c
6-14
芳基、c
6-14
芳基-c
1-12
烷基、5-14元杂芳基、5-14元杂芳基-c
1-12
烷基;
[0015]
每个ra、rb或rc相同或不同,彼此独立地选自h、卤素、cn、c
1-12
烷基、c
1-12
烷氧基、c
6-14
芳基、5-14元杂芳基。
[0016]
根据本发明的实施方案,每个r相同或不同,彼此独立地选自无取代、或任选被一个、两个或多个ra取代的c
1-10
烷基、c
6-10
芳基、c
6-10
芳基-c
1-10
烷基;
[0017]
根据本发明的实施方案,每个r'相同或不同,彼此独立地选自无取代、或任选被一个、两个或多个rb取代的c
6-10
芳基、c
6-10
芳基-c
1-10
烷基、5-10元杂芳基、5-10元杂芳基-c
1-10
烷基;
[0018]
根据本发明的实施方案,每个r”'相同或不同,彼此独立地选自无取代、或任选被一个、两个或多个rc取代的c
6-10
芳基、c
6-10
芳基-c
1-10
烷基、5-10元杂芳基、5-10元杂芳基-c
1-10
烷基;
[0019]
根据本发明的实施方案,每个ra、rb或rc相同或不同,彼此独立地选自h、cn、卤素、c
1-10
烷基、c
1-10
烷氧基、c
6-10
芳基、5-10元杂芳基。
[0020]
根据本发明的实施方案,每个r相同或不同,彼此独立地选自无取代、或任选被一个、两个或多个ra取代的c
1-10
烷基、苯基、苯基甲基、苯基乙基;
[0021]
根据本发明的实施方案,每个r'相同或不同,彼此独立地选自无取代、或任选被一个、两个或多个rb取代的苯基、苯基甲基、吡啶基、吡啶基甲基;
[0022]
根据本发明的实施方案,每个r”'相同或不同,彼此独立地选自无取代、或任选被
一个、两个或多个rc取代的苯基、苯基甲基、吡啶基、吡啶基甲基;;
[0023]
根据本发明的实施方案,每个ra、rb或rc相同或不同,彼此独立地选自h、cn、f、c
1-10
烷基、c
1-10
烷氧基。
[0024]
根据本发明的实施方案,每个r'、r”'相同或不同,彼此独立地选自下述基团中的一种:
[0025][0026]
根据本发明的实施方案,每个r相同或不同,彼此独立地选自下述基团中的一种:
[0027]
[0028]
其中*处为连接位点。
[0029]
优选地,所述有机荧光传感材料为一种花状的材料。
[0030]
本发明还提供所述有机荧光传感材料的制备方法,其包括以下步骤:
[0031]
(1)制备式(i)所示化合物;
[0032]
(2)制备式(ii)所示化合物;
[0033]
(3)将式(i)所示化合物和式(ii)所示化合物溶解在良溶剂中,然后加入不良溶剂,共组装得到所述有机荧光传感材料;或者将式(i)所示化合物溶解在良溶剂中,然后加入不良溶剂,然后加入式(ii)所示化合物的良溶剂溶液,共组装得到所述有机荧光传感材料;
[0034]
根据本发明的实施方案,所述化合物(i)的制备方法包括以下步骤(a1)-(a8):
[0035]
(a1)化合物a与rx反应得到化合物b;
[0036][0037]
(a2)化合物b与双戊酰二硼反应得到化合物c;
[0038][0039]
(a3)化合物d与化合物r
’‑
b(oh)2反应得到化合物e;
[0040][0041]
(a4)化合物e与双戊酰二硼反应得到化合物f;
[0042][0043]
(a5)化合物b与化合物f反应得到化合物g;
[0044][0045]
(a6)化合物g开环脱硫反应得到化合物h;
[0046][0047]
(a7)化合物h与原甲酸酯反应得到化合物i;
[0048][0049]
(a8)化合物i与化合物c反应得到式(i)所示化合物;
[0050][0051]
其中,x为卤素,n、r、r’具有上文所述的定义。
[0052]
根据本发明,所述步骤(a1)中,所述反应可以在溶剂中进行,所述溶剂可以为有机溶剂,例如二甲基亚砜、n,n-二甲基甲酰胺、n,n-二甲基乙酰胺;
[0053]
根据本发明,所述步骤(a1)中,所述反应的温度可以为-10~10℃,例如-5~5℃;
[0054]
根据本发明,所述步骤(a1)中,所述反应可以在碱作用下进行,所述碱可以为氢氧化钠、氢氧化钾、氢氧化锂或其水溶液,例如50%氢氧化钠的水溶液;
[0055]
根据本发明,所述步骤(a1)中,化合物a与rx的摩尔比为1:2-8,例如1:2-3。
[0056]
根据本发明,所述步骤(a2)中,所述反应在溶剂中进行,所述溶剂可以为有机溶剂,例如为醚类溶剂,如1,4-二氧六环、四氢呋喃;
[0057]
根据本发明,所述步骤(a2)中,化合物b与双戊酰二硼的摩尔比为1:1-8,例如1:2-5。
[0058]
根据本发明,所述步骤(a2)中,所述反应在催化剂体系中进行,所述催化剂体系包括钯催化剂和碱金属盐;所述钯催化剂可以为[1,1'-双(二苯基膦基)二茂铁]二氯化钯;所述碱金属盐可以为醋酸钾、碳酸钠、碳酸钾、碳酸铯;
[0059]
根据本发明,所述步骤(a2)中,所述化合物b、钯催化剂和碱金属盐的摩尔比可以为1:0.01-0.5:1-10,例如为1:0.05-0.2:2-10;
[0060]
根据本发明,所述步骤(a2)中,所述反应在惰性气体保护下进行,反应温度为70~90℃,反应时间为6~8小时。
[0061]
根据本发明,所述步骤(a3)中,所述反应在溶剂中进行,所述溶剂可以为有机溶剂,例如为醚类溶剂,如1,4-二氧六环、四氢呋喃;
[0062]
根据本发明,所述步骤(a3)中,所述化合物d与r
’‑
b(oh)2的摩尔比为1:1-8,例如为1:2-5;
[0063]
根据本发明,所述步骤(a3)中,所述反应在催化剂体系中进行,所述催化剂体系包括钯催化剂和碱金属盐;所述钯催化剂可以为四(三苯基膦)钯;所述碱金属盐可以为醋酸钾、碳酸钠、碳酸钾、碳酸铯;
[0064]
根据本发明,所述步骤(a3)中,所述化合物d、钯催化剂和碱金属盐的摩尔比可以为1:0.01-0.5:1-10,例如为1:0.05-0.2:2-8。
[0065]
根据本发明,所述步骤(a4)中,化合物e与双戊酰二硼的当量比为1:0.8-3,例如为1:1.2-1.5;
[0066]
根据本发明,所述步骤(a4)中,所述反应在催化剂体系中进行,所述催化剂体系包括钯催化剂和碱金属盐;所述钯催化剂可以为[1,1'-双(二苯基膦基)二茂铁]二氯化钯;所述碱金属盐可以为醋酸钾、碳酸钠、碳酸钾、碳酸铯;
[0067]
根据本发明,所述步骤(a4)中,所述化合物e、钯催化剂和碱金属盐的摩尔比可以为1:0.01-0.5:1-10,例如为1:0.05-0.2:2-10;
[0068]
根据本发明,所述步骤(a4)中,所述反应在惰性气体保护下进行,反应温度为70~80℃,反应时间为6~8小时。
[0069]
根据本发明,所述步骤(a5)中,所述反应在溶剂中进行,所述溶剂可以为有机溶剂,例如为醚类溶剂,如1,4-二氧六环、四氢呋喃;
[0070]
根据本发明,所述步骤(a5)中,所述反应在催化剂体系中进行,所述催化剂体系包括钯催化剂和碱金属盐;所述钯催化剂可以为四(三苯基膦)钯;所述碱金属盐可以为醋酸钾、碳酸钠、碳酸钾、碳酸铯;
[0071]
根据本发明,所述步骤(a5)中,所述化合物b与化合物f的摩尔比可以为1:0.8-1.2,例如为1:1;
[0072]
根据本发明,所述步骤(a5)中,所述化合物b、钯催化剂和碱金属盐的摩尔比可以为1:0.01-0.5:1-10,例如为1:0.05-0.2:2-8;
[0073]
根据本发明,所述步骤(a5)中,所述反应在惰性气体保护下进行,反应温度可以为70~90℃,反应时间可以为6~48小时。
[0074]
根据本发明,所述步骤(a6)中,所述反应在有机溶剂中进行,所述溶剂可以为四氢呋喃和乙醇的混合溶液,体积为1:1;
[0075]
根据本发明,所述步骤(a6)中,所述反应可以在还原剂存在下进行,所述还原剂可以为硼氢化钠、硼氢化钾、醋酸硼氢化钠、氰基硼氢化钠;所述反应可以在催化剂存在下进行,所述催化剂可以为三水氯化钴;
[0076]
根据本发明,所述步骤(a6)中,所述化合物g、还原剂和催化剂的摩尔比可以为1:1-10:0.01-0.5,例如为1:2-8:0.01-0.1;
[0077]
根据本发明,所述步骤(a6)中,所述反应的温度可以为60~80℃,反应时间可以为1~3小时。
[0078]
根据本发明,所述步骤(a7)中,所述反应在有机溶剂中进行,所述溶剂可以为四氢呋喃和甲醇的混合溶液,体积比为1:1;
[0079]
根据本发明,所述步骤(a7)中,所述反应可以在催化剂作用下进行,所述催化剂可以为氨基磺酸;
[0080]
根据本发明,所述步骤(a7)中,所述原甲酸酯可以为原甲酸三甲酯、原甲酸三乙酯
或原甲酸三异丙酯;
[0081]
根据本发明,所述步骤(a7)中,所述化合物h、原甲酸酯和催化剂的摩尔比可以为1:1-10:0.01-0.5,例如为1:1-5:0.01-0.1;
[0082]
根据本发明,所述步骤(a7)中,所述反应的温度可以为60~80℃,反应时间可以为1~3小时。
[0083]
根据本发明,所述步骤(a8)中,所述反应可以在有机溶剂中进行,所述有机溶剂可以为芳香类溶剂,如甲苯;
[0084]
根据本发明,所述步骤(a8)中,所述化合物i与化合物c的摩尔比可以为1:0.3-0.8,例如为1:0.5;
[0085]
根据本发明,所述步骤(a8)中,所述反应在催化剂体系中进行,所述催化剂体系包括钯催化剂和碱金属盐;所述钯催化剂可以为四(三苯基膦)钯;所述碱金属盐可以为醋酸钾、碳酸钠、碳酸钾、碳酸铯;
[0086]
根据本发明,所述步骤(a8)中,所述化合物i、钯催化剂和碱金属盐的摩尔比可以为1:0.01-0.5:1-10,例如为1:0.05-0.2:2-8;
[0087]
根据本发明,所述步骤(a8)中,所述反应在惰性气体保护下进行,反应温度可以为80~90℃,反应时间可以为12~48小时。
[0088]
根据本发明的实施方案,所述化合物(ii)的制备方法包括以下步骤(b1)-(b4):
[0089]
(b1)化合物d与化合物r
”’‑
b(oh)2反应得到化合物j;
[0090][0091]
(b2)化合物j与与双戊酰二硼反应得到化合物k;
[0092][0093]
(b3)化合物b与化合物k反应得到化合物l;
[0094][0095]
(b4)化合物l与化合物c反应得到式(ii)所示化合物;
[0096][0097]
其中,x为卤素,m、r、r
”’
具有上文所述的定义。
[0098]
根据本发明,所述步骤(b1)中,所述反应在溶剂中进行,所述溶剂可以为有机溶剂,例如为醚类溶剂,如1,4-二氧六环、四氢呋喃;
[0099]
根据本发明,所述步骤(b1)中,所述化合物d与r
”’‑
b(oh)2的摩尔比为1:1-8,例如为1:2-5;
[0100]
根据本发明,所述步骤(b1)中,所述反应在催化剂体系中进行,所述催化剂体系包括钯催化剂和碱金属盐;所述钯催化剂可以为四(三苯基膦)钯;所述碱金属盐可以为醋酸钾、碳酸钠、碳酸钾、碳酸铯;
[0101]
根据本发明,所述步骤(b1)中,所述化合物d、钯催化剂和碱金属盐的摩尔比可以为1:0.01-0.5:1-10,例如为1:0.05-0.2:2-8。
[0102]
根据本发明,所述步骤(b2)中,化合物j与双戊酰二硼的当量比为1:0.8-3,例如为1:1.2-1.5;
[0103]
根据本发明,所述步骤(b2)中,所述反应在催化剂体系中进行,所述催化剂体系包括钯催化剂和碱金属盐;所述钯催化剂可以为[1,1'-双(二苯基膦基)二茂铁]二氯化钯;所述碱金属盐可以为醋酸钾、碳酸钠、碳酸钾、碳酸铯;
[0104]
根据本发明,所述步骤(b2)中,所述化合物j、钯催化剂和碱金属盐的摩尔比可以为1:0.01-0.5:1-10,例如为1:0.05-0.2:2-10;
[0105]
根据本发明,所述步骤(b2)中,所述反应在惰性气体保护下进行,反应温度为70~80℃,反应时间为6~8小时。
[0106]
根据本发明,所述步骤(b3)中,所述反应在溶剂中进行,所述溶剂可以为有机溶剂,例如为醚类溶剂,如1,4-二氧六环、四氢呋喃;
[0107]
根据本发明,所述步骤(b3)中,所述反应在催化剂体系中进行,所述催化剂体系包括钯催化剂和碱金属盐;所述钯催化剂可以为四(三苯基膦)钯;所述碱金属盐可以为醋酸钾、碳酸钠、碳酸钾、碳酸铯;
[0108]
根据本发明,所述步骤(b3)中,所述化合物b与化合物k的摩尔比可以为1:0.8-1.2,例如为1:1;
[0109]
根据本发明,所述步骤(b3)中,所述化合物b、钯催化剂和碱金属盐的摩尔比可以为1:0.01-0.5:1-10,例如为1:0.05-0.2:2-8;
[0110]
根据本发明,所述步骤(b3)中,所述反应在惰性气体保护下进行,反应温度可以为70~90℃,反应时间可以为6~48小时。
[0111]
根据本发明,所述步骤(b4)中,所述反应可以在有机溶剂中进行,所述有机溶剂可以为芳香类溶剂,如甲苯;
[0112]
根据本发明,所述步骤(b4)中,所述化合物l与化合物c的摩尔比可以为1:0.3-0.8,例如为1:0.5;
[0113]
根据本发明,所述步骤(b4)中,所述反应在催化剂体系中进行,所述催化剂体系包括钯催化剂和碱金属盐;所述钯催化剂可以为四(三苯基膦)钯;所述碱金属盐可以为醋酸钾、碳酸钠、碳酸钾、碳酸铯;
[0114]
根据本发明,所述步骤(b4)中,所述化合物l、钯催化剂和碱金属盐的摩尔比可以为1:0.01-0.5:1-10,例如为1:0.05-0.2:2-8;
[0115]
根据本发明,所述步骤(b4)中,所述反应在惰性气体保护下进行,反应温度可以为80~90℃,反应时间可以为12~48小时。
[0116]
根据本发明的实施方案,步骤(3)中所述式(i)化合物和式(ii)化合物的摩尔比可以为(1~5000):1,例如为(1~2000):1,示例性为1:1、5:1、10:1、50:1、100:1、500:1。
[0117]
根据本发明,所述步骤(3)进一步包括:在共组装后,取出位于制备容器底部的有机荧光传感材料,再次置于不良溶剂中摇匀分散并反复洗涤。
[0118]
根据本发明,步骤(3)中所述良溶剂与不良溶剂的体积比(ml:ml)为1:2~1:15,优选1:3。
[0119]
根据本发明,步骤(3)中所述良溶剂为卤代烷烃类溶剂,例如选自二氯甲烷、氯仿或1,2-二氯乙烷中的至少一种。
[0120]
根据本发明,步骤(3)中所述不良溶剂为醇类溶剂或烷烃类溶剂,例如可以选自甲醇或正己烷中的至少一种。
[0121]
本发明还提供所述有机荧光传感材料在检测神经性毒剂中的用途。
[0122]
优选地,所述神经性毒剂选自沙林(sarin,gb)、梭曼(soman,gd)、塔崩(tabun,ga)、维埃克斯(vx)、氯磷酸二乙酯(dcp)、氟磷酸二异丙酯(dfp)和氰基磷酸二乙酯(dcnp)中的至少一种。
[0123]
本发明还提供一种检测神经性毒剂的方法,包括:将所述有机荧光传感材料与神经性毒剂接触,当有机荧光传感材料的荧光发生增强并且不恢复时,则为神经性毒剂;当有机荧光传感材料接触其他干扰物是发生快速恢复时,则为干扰物。
[0124]
作为实例,所述检测神经性毒剂的方法,包括以下步骤:将所述有机荧光传感材料涂覆在石英管的内壁,然后将石英管与泵相连接,用激发光源激发传感材料,将被检测的神经性毒剂蒸气吸入石英管内,通过荧光传感器检测荧光信号的差别,从而达到检测神经性毒剂的目的。
[0125]
根据本发明的实施方案,被检测的神经性毒剂浓度范围可以为0.01ppb到500ppm。
[0126]
有益效果
[0127]
1、本发明所述有机荧光传感材料为花状结构的有机半导体材料的多孔膜,其具有高比表面积,可以有效地检测神经性毒剂及其模拟物,具有很高的灵敏性,其检测的浓度可为0.01ppb-500ppm。
[0128]
2.本发明所述有机荧光传感材料对常见的有机溶剂(几千ppm)无响应或者出现溶剂化的荧光信号(荧光快速恢复),这种荧光变化与神经性毒剂的荧光信号(荧光增强)相反。因此本技术的方法可以避免其它有机溶剂的干扰,具有较高的检测专一性。
[0129]
3.本发明所述有机荧光传感材料通过使用对神经性毒剂不敏感的式(ii)所示化合物与式(i)所示化合物共组装,提高了单一使用式(i)化合物时检测的稳定性和抗干扰能力,克服了单一使用式(ii)化合物检测不灵敏的缺点。共组装的得到的材料具有比表面积
大,表面孔隙多等特征,有利于降低检出限。
附图说明
[0130]
图1实施例1中化合物1的核磁数据谱图。
[0131]
图2实施例2中化合物2的核磁数据谱图。
[0132]
图3实施例3中化合物3的核磁数据谱图。
[0133]
图4实施例4中化合物4的核磁数据谱图。
[0134]
图5实施例5中化合物5的核磁数据谱图。
[0135]
图6实施例6中化合物1和化合物3按照摩尔比50:1的比例进行共组装构筑而成的有机荧光传感材料的sem图像,其为花状结构的微米球。
[0136]
图7实施例7中化合物1和化合物4按照摩尔比50:1的比例进行共组装构筑而成的有机荧光传感材料的sem图像,其为花状结构的微米球。
[0137]
图8实施例8中化合物2和化合物4按照摩尔比50:1的比例进行共组装构筑而成的有机荧光传感材料的sem图像,其为花状结构的微米球。
[0138]
图9实施例9中化合物2和化合物5按照摩尔比50:1的比例进行共组装构筑而成的有机荧光传感材料的sem图像,其为花状结构的微米球。
[0139]
图10本发明化合物2和5的吸收荧光图,化合物2的荧光光谱与化合物5的吸收光谱有部分重叠,说明两者会发生荧光能量共振传递(实线和虚线分别为化合物2和化合物5的吸收荧光光谱)。
[0140]
图11实施例11中化合物1和化合物3按照摩尔比50:1的比例进行组装构筑而成的有机传感材料对氰基磷酸二乙酯(dcnp)气体的检测荧光曲线图,最低检测限为0.2ppm,荧光增强的响应。
[0141]
图12实施例12中化合物1和化合物4按照摩尔比50:1的比例进行组装构筑而成的有机传感材料对氰基磷酸二乙酯(dcnp)气体的检测荧光曲线图,最低检测限为0.2ppm,荧光增强的响应。
[0142]
图13实施例13中化合物2和化合物4按照摩尔比50:1的比例进行共组装构筑而成的有机传感材料对氰基磷酸二乙酯(dcnp)气体的检测荧光曲线图,最低检测限为50ppb,荧光增强的响应。
[0143]
图14实施例14中化合物2和化合物5按照摩尔比50:1的比例进行共组装构筑而成的有机传感材料对氰基磷酸二乙酯(dcnp)气体的检测荧光曲线图,最低检测限为50ppb,荧光增强的响应。
[0144]
图15实施例15中化合物2和化合物5按照摩尔比50:1的比例进行共组装构筑而成的有机传感材料对真实神经毒剂(沙林)的检测荧光曲线图,最低检测限为0.5ppm,荧光增强的响应。
[0145]
图16实施例16中化合物2和化合物5按照摩尔比50:1的比例进行共组装构筑而成的有机传感材料对干扰物(dcp)的检测荧光曲线图,呈现荧光淬灭的响应,可以有效排除其对检测的影响。
[0146]
图17实施例17中化合物2和化合物5按照摩尔比50:1的比例进行共组装构筑而成的有机传感材料对盐酸(hcl)的检测荧光曲线图,呈现荧光淬灭的响应,可以有效排除其对
检测的影响。
[0147]
图18实施例18中化合物2和化合物5按照摩尔比50:1的比例进行共组装构筑而成的有机传感材料对正己烷的检测荧光曲线图,呈现上升立即恢复,可以有效排除其对检测的影响。
[0148]
图19实施例19中化合物2和化合物5按照摩尔比50:1的比例进行共组装构筑而成的有机传感材料对乙醇的检测荧光曲线图,呈现上升立即恢复,可以有效排除其对检测的影响。
[0149]
图20实施例20中化合物2和化合物5按照摩尔比50:1的比例进行共组装构筑而成的有机传感材料对氯仿的检测荧光曲线图,呈现上升立即恢复,可以有效排除其对检测的影响。
[0150]
图21实施例21中化合物2和化合物5按照摩尔比50:1的比例进行共组装构筑而成的有机传感材料对甲醇的检测荧光曲线图,呈现上升立即恢复,可以有效排除其对检测的影响。
[0151]
图22对比例1中化合物2单独组装而成的有机传感材料的荧光曲线图。
[0152]
图23对比例1中化合物2单独组装而成的有机传感材料光照1小时后的荧光曲线图。
[0153]
图24对比例2中化合物2和化合物5按照摩尔比50:1的比例进行共组装构筑而成的有机传感材料在光照1小时后的荧光曲线图。
[0154]
图25对比例3中化合物4单独组装而成的有机传感材料对dcnp的检测荧光曲线图,说明本发明式(ii)化合物不能用于检测神经性毒剂。
[0155]
术语定义与说明
[0156]
除非另有说明,本技术说明书和权利要求书中记载的基团和术语定义,包括其作为实例的定义、示例性的定义、优选的定义、表格中记载的定义、实施例中具体化合物的定义等,可以彼此之间任意组合和结合。这样的组合和结合后的基团定义及化合物结构,应当被理解为本技术说明书和/或权利要求书记载的范围内。
[0157]
除非另有说明,本说明书和权利要求书记载的数值范围相当于至少记载了其中每一个具体的整数数值。例如,数值范围“1-12”相当于记载每一个整数数值即1、2、3、4、5、6、7、8、9、10、11、12。此外,当某些数值范围被定义为“数”时,应当理解为记载了该范围的两个端点、该范围内的每一个整数以及该范围内的每一个小数。例如,“0~10的数”应当理解为不仅记载了0、1、2、3、4、5、6、7、8、9和10的每一个整数,还至少记载了其中每一个整数分别与0.1、0.2、0.3、0.4、0.5、0.6、0.7、0.8、0.9的和。
[0158]
应当理解,本文在描述1、2个或多个中,“多个”应当是指大于2,例如大于等于3的整数,例如3、4、5、6、7、8、9或10。
[0159]
术语“卤素”表示氟、氯、溴和碘。
[0160]
术语“c
1-12
烷基”应理解为表示具有1~12个碳原子的直链或支链饱和一价烃基。例如,“c
1-12
烷基”表示具有1、2、3、4、5、6、7、8、9、10、11或12个碳原子的直链和支链烷基,“c
1-6
烷基”表示具有1、2、3、4、5或6个碳原子的直链和支链烷基。所述烷基是例如甲基、乙基、丙基、丁基、戊基、己基、异丙基、异丁基、仲丁基、叔丁基、异戊基、2-甲基丁基、1-甲基丁基、1-乙基丙基、1,2-二甲基丙基、新戊基、1,1-二甲基丙基、4-甲基戊基、3-甲基戊基、2-甲
基戊基、1-甲基戊基、2-乙基丁基、1-乙基丁基、3,3-二甲基丁基、2,2-二甲基丁基、1,1-二甲基丁基、2,3-二甲基丁基、1,3-二甲基丁基或1,2-二甲基丁基等或它们的异构体。
[0161]
术语“c
6-14
芳基”应理解为优选表示具有6~14个碳原子的一价芳香性或部分芳香性的单环、二环(如稠环、桥环、螺环)或三环烃环,其可以是单芳族环或稠合在一起的多芳族环,术语“c
6-14
芳基”应理解为优选表示具有6、7、8、9、10、11、12、13或14个碳原子的一价芳香性或部分芳香性的单环、双环或三环烃环(“c
6-14
芳基”),特别是具有6个碳原子的环(“c6芳基”),例如苯基;或联苯基,或者是具有9个碳原子的环(“c9芳基”),例如茚满基或茚基,或者是具有10个碳原子的环(“c
10
芳基”),例如四氢化萘基、二氢萘基或萘基,或者是具有13个碳原子的环(“c
13
芳基”),例如芴基,或者是具有14个碳原子的环(“c
14
芳基”),例如蒽基。当所述c
6-20
芳基被取代时,其可以为单取代或者多取代。并且,对其取代位点没有限制,例如可以为邻位、对位或间位取代。
[0162]
术语“5-14元杂芳基”应理解为包括这样的一价单环、二环(如稠环、桥环、螺环)或三环芳族环系:其具有5~14个环原子且包含1-5个独立选自n、o和s的杂原子。术语“5-14元杂芳基”应理解为包括这样的一价单环、双环或三环芳族环系:其具有5、6、7、8、9、10、11、12、13或14个环原子,特别是5或6或9或10个碳原子,且其包含1-5个,优选1-3各独立选自n、o和s的杂原子并且,另外在每一种情况下可为苯并稠合的。“杂芳基”还指其中杂芳族环与一个或多个芳基、脂环族或杂环基环稠合的基团,其中所述连接的根基或点在杂芳族环上。。当所述5-14元杂芳基与其它基团相连构成本发明的化合物时,可以为5-14元杂芳基环上的碳原子与其它基团相连,也可以为5-14元杂芳基环上的杂原子与其它基团相连。当所述5-14元杂芳基被取代时,其可以为单取代或者多取代。并且,对其取代位点没有限制,例如可以为杂芳基环上与碳原子相连的氢被取代,或者杂芳基环上与杂原子相连的氢被取代。
具体实施方式
[0163]
下文将结合具体实施例对本发明的技术方案做更进一步的详细说明。应当理解,下列实施例仅为示例性地说明和解释本发明,而不应被解释为对本发明保护范围的限制。凡基于本发明上述内容所实现的技术均涵盖在本发明旨在保护的范围内。
[0164]
除非另有说明,以下实施例中使用的原料和试剂均为市售商品,或者可以通过已知方法制备。
[0165]
由于神经性毒剂(如gb、gd、ga)毒性很强,氯磷酸二乙酯(dcp)是一种常见的模拟物,由于其很容易吸水分解产生盐酸,所以其不适合作为神经性毒剂模拟物,因此采用氰基磷酸二乙酯(dcnp)作为神经性毒剂的模拟物,其与真实的化学战剂具有相同的反应活性,并且实际测试真实毒剂的响应。
[0166]
神经性毒剂模拟物真实神经性毒剂
[0167]
实施例1
[0168]
制备如下化合物1,其制备方法如下所示:
[0169][0170]
(1)将10克的2,7-二溴芴和11.5克的溴己烷溶于40毫升的二甲基亚砜(dmso)溶液中,缓慢加入10毫升的50%的氢氧化钠水溶液,在室温下反应3h后,加入100毫升的水淬灭反应,然后用乙酸乙酯萃取三遍,最后将得到的物质通过柱层析得到产物(tm-1)。
[0171]
(2)取步骤(1)得到的产物5克,加入40毫升1,4-二氧六环溶液中,加入3当量的双戊酰二硼、8当量的醋酸钾、10%当量的[1,1'-双(二苯基膦基)二茂铁]二氯化钯在80℃氩气保护下,反应6小时后,通过柱层析得到产物(tm-2)。
[0172]
(3)取3.6克的异丁氧基苯硼醇和4.12克的二溴苯并噻二唑,加入30毫升1,4-二氧六环溶液和5毫升水混合溶液中,加入10%当量的四(三苯基膦)钯、3当量的碳酸铯在80℃氩气保护下,反应6小时后,通过柱层析得到产物(tm-3)。
[0173]
(4)取步骤(3)得到的产物2克加入40毫升1,4-二氧六环溶液中,加入1.5当量的双戊酰二硼、4当量的醋酸钾、10%当量的[1,1'-双(二苯基膦基)二茂铁]二氯化钯在80℃氩气保护下,反应6小时后,通过柱层析得到产物(tm-4)。
[0174]
(5)分别取步骤(1)和步骤(4)的产物1mmol加入到30毫升的1,4-二氧六环溶液和5毫升水的混合溶液中,加入10%当量的四(三苯基膦)钯、3当量的碳酸铯在80℃氩气保护下,反应6小时后,通过柱层析得到产物(tm-5)。
[0175]
(6)取步骤(5)得到的产物1.35克加入到10毫升四氢呋喃和10毫升乙醇的混合溶液中,然后加入5当量的硼氢化钠和5%三水合氯化钴,回流反应2h后,通过柱层析得到产物(tm-6)。
[0176]
(7)取步骤(6)得到的产物0.67克加入到5毫升四氢呋喃和5毫升甲醇混合溶液中,然后加入2当量的原甲酸三乙酯和5%的氨基磺酸,反应2h后,通过柱层析得到产物(tm-7)。
[0177]
(8)分别取步骤(2)和步骤(7)的产物0.4mmol和0.8mmol加入到20毫升的甲苯溶液和4毫升水的混合溶液中,加入10%当量的四(三苯基膦)钯、5当量的碳酸铯在95℃氩气保护下,反应过夜后,通过柱层析得到产物1。其核磁共振数据图如图1所示。
[0178]
实施例2
[0179]
制备如下化合物2,其制备方法如下所示:
[0180][0181]
(1)将10克的2,7-二溴芴和7.2克的溴乙烷溶于40毫升的二甲基亚砜(dmso)溶液中,缓慢加入10毫升的50%的氢氧化钠水溶液,,在室温下反应3h后,加入100毫升的水淬灭反应,然后用乙酸乙酯萃取三遍,最后将得到的物质通过柱层析得到产物(tm-1)。
[0182]
(2)取步骤(1)得到的产物5克,加入40毫升1,4-二氧六环溶液中,加入3当量的双戊酰二硼、8当量的醋酸钾、10%当量的[1,1'-双(二苯基膦基)二茂铁]二氯化钯在80℃氩气保护下,反应6小时后,通过柱层析得到产物(tm-2)。
[0183]
(3)取3.6克的异丁氧基苯硼醇和4.12克的二溴苯并噻二唑,加入30毫升1,4-二氧六环溶液和5毫升水混合溶液中,加入10%当量的四(三苯基膦)钯、3当量的碳酸铯在80℃氩气保护下,反应6小时后,通过柱层析得到产物(tm-3)。
[0184]
(4)取步骤(3)得到的产物2克加入40毫升1,4-二氧六环溶液中,加入1.5当量的双戊酰二硼、4当量的醋酸钾、10%当量的[1,1'-双(二苯基膦基)二茂铁]二氯化钯在80℃氩气保护下,反应6小时后,通过柱层析得到产物(tm-4)。
[0185]
(5)分别取步骤(1)和步骤(4)的产物1mmol加入到30毫升的1,4-二氧六环溶液和5毫升水的混合溶液中,加入10%当量的四(三苯基膦)钯、3当量的碳酸铯在80℃氩气保护下,反应6小时后,通过柱层析得到产物(tm-5)。
[0186]
(6)取步骤(5)得到的产物1.35克加入到10毫升四氢呋喃和10毫升乙醇的混合溶液中,然后加入5当量的硼氢化钠和5%三水合氯化钴,回流反应2h后,通过柱层析得到产物(tm-6)。
[0187]
(7)取步骤(6)得到的产物0.67克加入到5毫升四氢呋喃和5毫升甲醇混合溶液中,然后加入2当量的原甲酸三乙酯和5%的氨基磺酸,反应2h后,通过柱层析得到产物(tm-7)。
[0188]
(8)分别取步骤(2)和步骤(7)的产物0.4mmol和0.8mmol加入到20毫升的甲苯溶液和4毫升水的混合溶液中,加入10%当量的四(三苯基膦)钯、5当量的碳酸铯在95℃氩气保护下,反应过夜后,通过柱层析得到产物2。其核磁共振数据图如图2所示。
[0189]
实施例3
[0190]
制备如下化合物3,其制备方法如下所示:
[0191][0192]
(1)将10克的2,7-二溴芴和11.5克的溴己烷溶于40毫升的二甲基亚砜(dmso)溶液中,缓慢加入10毫升的50%的氢氧化钠水溶液,,在室温下反应3h后,加入100毫升的水淬灭反应,然后用乙酸乙酯萃取三遍,最后将得到的物质通过柱层析得到产物(tm-1)。
[0193]
(2)取步骤(1)得到的产物5克,加入40毫升1,4-二氧六环溶液中,加入3当量的双戊酰二硼、8当量的醋酸钾、10%当量的[1,1'-双(二苯基膦基)二茂铁]二氯化钯在80℃氩气保护下,反应6小时后,通过柱层析得到产物(tm-2)。
[0194]
(3)取3.6克的异丁氧基苯硼醇和4.12克的二溴苯并噻二唑,加入30毫升1,4-二氧六环溶液和5毫升水混合溶液中,加入10%当量的四(三苯基膦)钯、3当量的碳酸铯在80℃氩气保护下,反应6小时后,通过柱层析得到产物(tm-3)。
[0195]
(4)取步骤(3)得到的产物2克加入40毫升1,4-二氧六环溶液中,加入1.5当量的双戊酰二硼、4当量的醋酸钾、10%当量的[1,1'-双(二苯基膦基)二茂铁]二氯化钯在80℃氩气保护下,反应6小时后,通过柱层析得到产物(tm-4)。
[0196]
(5)分别取步骤(1)和步骤(4)的产物1mmol加入到30毫升的1,4-二氧六环溶液和5毫升水的混合溶液中,加入10%当量的四(三苯基膦)钯、3当量的碳酸铯在80℃氩气保护下,反应6小时后,通过柱层析得到产物(tm-5)。
[0197]
(6)分别取步骤(2)和步骤(5)的产物1.5mmol和3.1mmol加入到20毫升的甲苯溶液和4毫升水的混合溶液中,加入10%当量的四(三苯基膦)钯、3当量的碳酸铯在80℃氩气保护下,反应过夜后,通过柱层析得到产物3。其核磁共振数据图如图3所示。
[0198]
实施例4
[0199]
制备如下化合物4,其制备方法如下所示:
[0200][0201]
(1)将10克的2,7-二溴芴和7.2克的溴乙烷溶于40毫升的二甲基亚砜(dmso)溶液中,缓慢加入10毫升的50%的氢氧化钠水溶液,,在室温下反应3h后,加入100毫升的水淬灭反应,然后用乙酸乙酯萃取三遍,最后将得到的物质通过柱层析得到产物(tm-1)。
[0202]
(2)取步骤(1)得到的产物5克,加入40毫升1,4-二氧六环溶液中,加入3当量的双戊酰二硼、8当量的醋酸钾、10%当量的[1,1'-双(二苯基膦基)二茂铁]二氯化钯在80℃氩气保护下,反应6小时后,通过柱层析得到产物(tm-2)。
[0203]
(3)取3.6克的异丁氧基苯硼醇和4.12克的二溴苯并噻二唑,加入30毫升1,4-二氧六环溶液和5毫升水混合溶液中,加入10%当量的四(三苯基膦)钯、3当量的碳酸铯在80℃氩气保护下,反应6小时后,通过柱层析得到产物(tm-3)。
[0204]
(4)取步骤(3)得到的产物2克加入40毫升1,4-二氧六环溶液中,加入1.5当量的双戊酰二硼、4当量的醋酸钾、10%当量的[1,1'-双(二苯基膦基)二茂铁]二氯化钯在80℃氩气保护下,反应6小时后,通过柱层析得到产物(tm-4)。
[0205]
(5)分别取步骤(1)和步骤(4)的产物1mmol加入到30毫升的1,4-二氧六环溶液和5毫升水的混合溶液中,加入10%当量的四(三苯基膦)钯、3当量的碳酸铯在80℃氩气保护下,反应6小时后,通过柱层析得到产物(tm-5)。
[0206]
(6)分别取步骤(2)和步骤(5)的产物1.5mmol和3.1mmol加入到20毫升的甲苯溶液和4毫升水的混合溶液中,加入10%当量的四(三苯基膦)钯、3当量的碳酸铯在80℃氩气保护下,反应过夜后,通过柱层析得到产物4。其核磁共振数据图如图4所示。
[0207]
实施例5
[0208][0209]
(1)将10克的2,7-二溴芴和10克的溴丁烷溶于40毫升的二甲基亚砜(dmso)溶液中,缓慢加入10毫升的50%的氢氧化钠水溶液,,在室温下反应3h后,加入100毫升的水淬灭反应,然后用乙酸乙酯萃取三遍,最后将得到的物质通过柱层析得到产物(tm-1)。
[0210]
(2)取步骤(1)得到的产物5克,加入40毫升1,4-二氧六环溶液中,加入3当量的双戊酰二硼、8当量的醋酸钾、10%当量的[1,1'-双(二苯基膦基)二茂铁]二氯化钯在80℃氩气保护下,反应6小时后,通过柱层析得到产物(tm-2)。
[0211]
(3)取3.6克的异丁基苯硼醇和4.12克的二溴苯并噻二唑,加入30毫升1,4-二氧六环溶液和5毫升水混合溶液中,加入10%当量的四(三苯基膦)钯、3当量的碳酸铯在80℃氩气保护下,反应6小时后,通过柱层析得到产物(tm-3)。
[0212]
(4)取步骤(3)得到的产物2克加入40毫升1,4-二氧六环溶液中,加入1.5当量的双戊酰二硼、4当量的醋酸钾、10%当量的[1,1'-双(二苯基膦基)二茂铁]二氯化钯在80℃氩气保护下,反应6小时后,通过柱层析得到产物(tm-4)。
[0213]
(5)分别取步骤(1)和步骤(4)的产物1mmol加入到30毫升的1,4-二氧六环溶液和5毫升水的混合溶液中,加入10%当量的四(三苯基膦)钯、3当量的碳酸铯在80℃氩气保护下,反应6小时后,通过柱层析得到产物(tm-5)。
[0214]
(6)分别取步骤(2)和步骤(5)的产物1.5mmol和3.1mmol加入到20毫升的甲苯溶液和4毫升水的混合溶液中,加入10%当量的四(三苯基膦)钯、3当量的碳酸铯在80℃氩气保护下,反应过夜后,通过柱层析得到产物5。其核磁共振数据图如图5所示。
[0215]
实施例6
[0216]
分别将化合物1和化合物3溶解在良溶剂中分别得到浓度为5mg/ml的溶液。分别将化合物1和化合物3按照摩尔比10:1、20:1、50:1、100:1、500:1混合在一起,然后加入一定量的不良溶剂进行组装。所述良溶剂为二氯甲烷(将二氯甲烷替换为氯仿或1,2-二氯乙烷可以得到相同的结果),所述不良溶剂为正己烷。良溶剂与不良溶剂的体积比为1:10;静置得到有机荧光传感材料的悬浮液。
[0217]
实施例7
[0218]
分别将化合物1和化合物4溶解在良溶剂中分别得到浓度为5mg/ml的溶液。分别将化合物1和化合物4按照摩尔比10:1、20:1、50:1、100:1、500:1混合在一起,然后加入一定量的不良溶剂进行共组装。所述良溶剂为二氯甲烷(将二氯甲烷替换为氯仿或1,2-二氯乙烷可以得到相同的结果),所述不良溶剂为正己烷。良溶剂与不良溶剂的体积比为1:10;静置得到有机荧光传感材料的悬浮液。
[0219]
实施例8
[0220]
分别将化合物2和化合物4溶解在良溶剂中分别得到浓度为5mg/ml的溶液。分别将化合物2和化合物4按照摩尔比10:1、20:1、50:1、100:1、200:1、500:1混合在一起,然后加入一定量的不良溶剂进行共组装。所述良溶剂为二氯甲烷(将二氯甲烷替换为氯仿或1,2-二氯乙烷可以得到相同的结果),所述不良溶剂为甲醇(将甲醇替换为正己烷可以得到相同的结果),良溶剂与不良溶剂的体积比为1:10;静置,得到有机荧光传感材料的悬浮液。
[0221]
实施例9
[0222]
分别将化合物2和化合物5溶解在良溶剂中分别得到浓度为5mg/ml的溶液。分别将化合物2和化合物5按照摩尔比10:1、20:1、50:1、100:1、200:1、500:1混合在一起,然后加入一定量的不良溶剂进行共组装。所述良溶剂为二氯甲烷(将二氯甲烷替换为氯仿或1,2-二氯乙烷可以得到相同的结果),所述不良溶剂为甲醇(将甲醇替换为正己烷可以得到相同的结果),良溶剂与不良溶剂的体积比为1:10;静置,得到有机荧光传感材料的悬浮液。
[0223]
实施例10
[0224]
将实施例6、7、8和9制备好的悬浊液分别用移液枪取出容器底部的样品(2微升)并置于干净的硅片表面,待溶液挥发干净后将其放置于离子溅射机中(leica),抽真空到真空度为10-5
pa后开始表面溅射金属铂颗粒120s。取出硅片并将其置于扫描电镜(hitachi s4800)观察其形貌。电镜图表明材料是分子通过排列形成一种花状结构的球,其具有多孔结构,这为传感性能提供了足够的比表面积有利于气体在其内部扩散。
[0225]
实施例11
[0226]
将实施例6得到的悬浮液静置20小时后,将花状结构材料涂覆在石英管内(涂覆长度为2.5厘米到3厘米),石英管与一台微型流量泵连接,抽速为150ml/min。使用385纳米激发光源激发所述膜。用40ml的玻璃瓶稀释得到不同浓度的dcnp蒸汽,用泵将dcnp抽入到材料表面,检测结果都表现为出现了明显的荧光增强。如图11所示,浓度为0.2ppm的氰基磷酸二乙酯蒸汽可以产生荧光增强响应。
[0227]
实施例12
[0228]
将实施例7得到的悬浮液静置20小时后,将花状结构材料涂覆在石英管内,石英管与一台微型流量泵连接,抽速为150ml/min。使用385纳米激发光源激发所述膜。用40ml的玻璃瓶稀释得到不同浓度的dcnp蒸汽,用泵将dcnp抽入到材料表面,检测结果都表现为出现了明显的荧光增强。如图12所示,浓度为0.2ppm的氰基磷酸二乙酯蒸汽可以产生荧光增强响应。
[0229]
实施例13
[0230]
将实施例8得到的悬浮液静置20小时后,将花状结构材料涂覆在石英管内,石英管与一台微型流量泵连接,抽速为150ml/min。使用385纳米激发光源激发所述膜。用40ml的玻璃瓶稀释得到不同浓度的dcnp蒸汽,用泵将dcnp抽入到材料表面,检测结果都表现为出现了明显的荧光增强。如图13所示,浓度为50ppb的氰基磷酸二乙酯蒸汽可以产生荧光增强响应。
[0231]
实施例14
[0232]
将实施例9得到的悬浮液静置20小时后,将花状结构材料涂覆在石英管内,石英管与一台微型流量泵连接,抽速为150ml/min。使用385纳米激发光源激发所述膜。用40ml的玻璃瓶稀释得到不同浓度的dcnp蒸汽,用泵将dcnp抽入到材料表面,检测结果都表现为出现了明显的荧光增强。如图14所示,浓度为50ppb的氰基磷酸二乙酯蒸汽可以产生荧光增强响应。
[0233]
实施例15
[0234]
将实施例9得到的悬浮液静置20小时后,将花状结构材料涂覆在石英管内,石英管与一台微型流量泵连接,抽速为150ml/min。使用385纳米激发光源激发所述膜。用40ml的玻璃瓶稀释得到不同浓度的真实神经性毒剂(沙林),用泵将沙林抽入到材料表面,检测结果都表现为出现了明显的荧光增强,与模拟物产生相同的响应。如图15所示,浓度为0.5ppm的沙林蒸汽可以产生荧光增强响应。
[0235]
实施例16
[0236]
采用实施例11同样的方法,将实施例9得到的悬浮液静置20小时后,将花状结构材料涂覆在石英管内,石英管与一台微型流量泵连接,抽速为150ml/min。使用385纳米激发光源激发所述膜。只是将检测物替换为0.05ppm、0.1ppm、0.3ppm的氯磷酸二乙酯(dcp)蒸汽,检测结果表现出所述花状结构材料对dcp的响应是荧光淬灭的响应。说明这种混合材料可以排除dcp的干扰,而减少实际检测过程中的误判,检测结果如图16。
[0237]
实施例17
[0238]
采用实施例11同样的方法,将实施例9得到的悬浮液静置20小时后,将花状结构材料涂覆在石英管内,石英管与一台微型流量泵连接,抽速为150ml/min。使用385纳米激发光源激发所述膜。只是将检测物替换为50ppm、100ppm、300ppm的盐酸(hcl)蒸汽,检测结果表现出所述花状结构材料对hcl的响应是荧光淬灭的响应。说明这种混合材料可以排除hcl的干扰,而减少实际检测过程中的误判,检测结果如图17。
[0239]
实施例18
[0240]
采用实施例11同样的方法,将实施例9得到的悬浮液静置20小时后,将花状结构材料涂覆在石英管内,石英管与一台微型流量泵连接,抽速为150ml/min。使用385纳米激发光
源激发所述膜。只是将检测物替换为50ppm、240ppm、480ppm的丙酮蒸汽,检测结果表现出所述花状结构材料对丙酮产生一种溶剂化响应(即快速恢复响应),这样在痕量检测检测神经性毒剂时,丙酮不会对检测产生干扰,检测结果如图18。
[0241]
实施例19
[0242]
采用实施例11同样的方法,将实施例9得到的悬浮液静置20小时后,将花状结构材料涂覆在石英管内,石英管与一台微型流量泵连接,抽速为150ml/min。使用385纳米激发光源激发所述膜。只是将检测物替换为58ppm、120ppm、580ppm的乙醇蒸汽,检测结果表现出所述花状结构材料对乙醇产生一种溶剂化响应(即快速恢复响应),这样在痕量检测检测神经性毒剂时,乙醇不会对检测产生干扰,检测结果如图19。
[0243]
实施例20
[0244]
采用实施例11同样的方法,将实施例9得到的悬浮液静置20小时后,将花状结构材料涂覆在石英管内,石英管与一台微型流量泵连接,抽速为150ml/min。使用385纳米激发光源激发所述膜。只是将检测物替换为60ppm、340ppm、680ppm的二氯甲烷蒸汽,检测结果表现出所述多孔膜对二氯甲烷的响应是一种溶剂化响应(即快速恢复响应),这样在痕量检测检测神经性毒剂时,二氯甲烷不会对检测产生干扰,检测结果如图20。
[0245]
实施例21
[0246]
采用实施例11同样的方法,将实施例9得到的悬浮液静置20小时后,将花状结构材料涂覆在石英管内,石英管与一台微型流量泵连接,抽速为150ml/min。使用385纳米激发光源激发所述膜。只是将检测物替换为20ppm、100ppm、200ppm的四氢呋喃蒸汽,检测结果表现出所述多孔膜对四氢呋喃的响应是一种溶剂化响应(即快速恢复响应),这样在痕量检测或区分检测化学战剂(神经性毒剂和糜烂性毒剂)时,四氢呋喃不会对检测产生干扰,检测结果如图21。
[0247]
对比例1
[0248]
将化合物2溶解在良溶剂中得到实施例9中相同浓度的溶液。将化合物2的良溶剂加入一定量的不良溶剂进行自组装。所述良溶剂为二氯甲烷(将二氯甲烷替换为氯仿或1,2-二氯乙烷可以得到相同的结果),所述不良溶剂为正己烷(将甲醇替换为正己烷得到相同的结果),良溶剂与不良溶剂的体积比为1:10,然后用一个led光源(强度为0.041mw/cm2)激发所得到的纳米线,用荧光仪监控其荧光强度的变化,发现纳米线的荧光强度在光照1小时后衰减了60%,表明其荧光稳定性较差,实验结果如图22;并且在光照后,材料对水变得更加敏感,实验结果如图23。
[0249]
对比例2
[0250]
采用实施例11同样的方法,将实施例9得到的悬浮液静置20小时后,将花状结构材料涂覆在石英管内,石英管与一台微型流量泵连接,抽速为150ml/min。使用385纳米激发光源激发所述膜。将材料光照一小时后,检测其对水的响应,检测结果表现出所述花状结构材料对水产生较弱的响应,说明材料在光照过程中具有较强的抗氧化作用,这样在痕量检测神经性毒剂时,水不会对检测产生干扰,检测结果如图24。
[0251]
对比例3
[0252]
将化合物4溶解在良溶剂中得到分别得到浓度为5mg/ml的溶液,然后加入一定量的不良溶剂进行共组装。所述良溶剂为氯仿(将氯仿替换为二氯甲烷可以得到相同的结
果),所述不良溶剂为甲醇。采用实施例11同样的方法,将得到的悬浮液静置20小时后,将材料涂覆在石英管内,石英管与一台微型流量泵连接,抽速为150ml/min。使用385纳米激发光源激发所述膜。将材料光照一小时后,检测其对dcnp的响应,检测结果表现出所述由式(ii)化合物组装得到的材料对dcnp几乎没有响应,这说明这个材料是不能用于检测神经性毒剂,检测结果如图25。
[0253]
以上,对本发明的实施方式进行了说明。但是,本发明不限定于上述实施方式。凡在本发明的精神和原则之内,所做的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1