一种高选择性磁性染料吸附剂及其制备方法与流程
本发明涉及一种高选择性磁性染料吸附剂及其制备方法,属于功能高分子材料合成领域。
背景技术:
染料废弃物作为水污染的重要组成部分,已经成为生态系统的致命威胁之一。痕量有机染料的存在足以造成水体明显着色,从而降低太阳光在水中的透过率,并进一步对水体生态系统包含光合作用等在内的正常运转造成影响。更为严重的是,由于大多数有机染料含有芳香烃等复杂结构,其被生物降解的过程相对缓慢,大量具有毒性、致病性和致畸性的有机染料,特别是偶氮类染料,会通过食物链在生物体内富集。考虑到上述严峻形势,有机染料水体污染的治理迫在眉睫。
近数十年中,研究者们提出并发展了包括吸附、光催化分解、氧化、膜分离、离子交换以及生物处理在内的众多技术手段来处理水体污染。其中,具有安全、经济、易操作等优势的吸附方法是目前应用范围最为广泛的一类分离有毒物质的手段。然而,传统吸附剂回收难度高、吸附容量有限、吸附速率低下和选择性较弱等缺点仍然限制着大多数吸附方法的广泛实际使用。如,中国专利文献cn105502564a公开了一种选择性去除废水中含磺酸基团染料的吸附剂及其制备方法,利用纳米片状的碱式硝酸铜为吸附剂来去除废水中的含有磺酸基团的染料;虽然该吸附剂对含有磺酸基团染料具有选择性和很高的吸附量,但该吸附剂回收过程繁琐,回收成本较高,单一的选择性无法对废水中的其它种类染料进行有效吸附脱除,同时其可能对水体造成二次污染。又如,中国专利文献cn105664885a公开利用ph调节的磁性金属-有机骨架材料去除染料的方法,采用共沉淀法制备fe3o4,然后将其表面进行改性,生成硅烷化的磁性sio2,并与金属-有机骨架材料进行结合,其中金属-有机骨架材料由铝和氨基对苯二甲酸构建而成;通过调节ph值,使得制备的磁性金属-有机骨架材料作为一种新型的吸附剂,可以吸附不同种类的染料;该发明所制备的磁性金属-有机骨架材料具有比表面积大、较高的吸附容量、可磁分离再生利用等优点,但该发明吸附量有限,吸附速率欠佳。
技术实现要素:
针对现有技术的不足,本发明提供一种高选择性磁性染料吸附剂,该吸附剂易通过磁场快速回收,吸附容量较高,吸附速率快,能够对污水中的阴/阳离子型有机染料进行选择性的高效吸附脱除。
术语说明:
lysine:赖氨酸的英文名称;
pgma:聚甲基丙烯酸缩水甘油酯的简称。
本发明的技术方案如下:
一种高选择性磁性染料吸附剂,具有式ⅰ所示的结构,所述吸附剂是以fe3o4表面包覆sio2为核,核上接枝有lysine改性的pgma;
式ⅰ中,100≤n≤300。
根据本发明优选的,所述吸附剂的ph适用范围为2-11,接枝率为48-89%。
上述高选择性磁性染料吸附剂的制备方法,包括步骤:
(1)磁性纳米fe3o4的制备;
(2)纳米fe3o4-sio2的制备;
(3)纳米fe3o4-sio2的表面氨基改性;
(4)纳米fe3o4-sio2-nh2的溴代改性;
(5)纳米fe3o4-sio2-pgma的制备;
(6)纳米fe3o4-sio2-pgma的lysine改性:
将步骤(5)制备的纳米fe3o4-sio2-pgma分散于去离子水和1,4-二氧六环的混合液中,加入na2co3和赖氨酸(lysine),分散均匀,惰性气体、搅拌条件下,于50-80℃搅拌反应6-24h,经外加磁场分离、洗涤、干燥,即得高选择性磁性染料吸附剂。
根据本发明优选的,步骤(1)中所述磁性纳米fe3o4的制备包括步骤:
将fecl3·6h2o和无水naac均匀分散在乙二醇中,于180-210℃水热反应15-20h,经外加磁场分离、洗涤、干燥,得到磁性纳米fe3o4。
优选的,所述fecl3·6h2o与无水naac的质量比为1:2-1:3,fecl3·6h2o与乙二醇的质量比为1:30-1:45。
优选的,所述水热反应温度为190-200℃;所述洗涤条件为:无水乙醇洗涤5-10次;所述干燥条件为:室温真空干燥。
根据本发明优选的,步骤(2)中所述纳米fe3o4-sio2的制备包括步骤:
将步骤(1)得到的磁性纳米fe3o4分散在无水乙醇中,加入质量浓度为25-28%的氨水和正硅酸四乙酯,分散均匀,于25-60℃搅拌反应12-30h,经外加磁场分离、洗涤、干燥,得到纳米fe3o4-sio2。
优选的,所述磁性纳米fe3o4与正硅酸四乙酯的质量比为1:15-1:95,氨水与正硅酸四乙酯的质量比为0.1:1-1.2:1,磁性纳米fe3o4与无水乙醇的质量比为1:2000-1:2500。
优选的,所述反应温度为50℃,反应时间为24h-25h。
优选的,所述洗涤条件为:无水乙醇和去离子水交替洗涤;所述干燥条件为:室温真空干燥。
根据本发明优选的,步骤(3)中所述纳米fe3o4-sio2的表面氨基改性,包括步骤:
将步骤(2)制备的纳米fe3o4-sio2均匀分散在无水乙醇中,加入3-氨丙基三乙氧基硅烷偶联剂(kh550),分散均匀,于惰性气体保护下,70-90°搅拌反应1-5h,经外加磁场分离、洗涤、干燥,得到纳米fe3o4-sio2-nh2。
优选的,所述纳米fe3o4-sio2与3-氨丙基三乙氧基硅烷偶联剂的质量比为1:10-1:30,纳米fe3o4-sio2与无水乙醇的质量比为1:2000-1:2500。
优选的,所述惰性气体为氮气、氩气或氦气中的一种。
优选的,所述反应温度为85℃;所述洗涤条件为:无水乙醇洗涤5-10次;所述干燥条件为:室温真空干燥。
根据本发明优选的,步骤(4)中所述纳米fe3o4-sio2-nh2的溴代改性,包括步骤:
将步骤(3)制备的纳米fe3o4-sio2-nh2均匀分散在二氯甲烷中,加入三乙胺作为缚酸剂,冰浴条件下逐滴滴加2-溴异丁酰溴,分散均匀,室温下搅拌反应6-30h,经外加磁场分离、洗涤、干燥,得到纳米fe3o4-sio2-br。
优选的,所述纳米fe3o4-sio2-nh2与2-溴异丁酰溴的质量比为1:5-1:10,三乙胺与2-溴异丁酰溴的摩尔比为3:1-1:1,纳米fe3o4-sio2-nh2与二氯甲烷的质量比为1:3500-1:4200。
优选的,所述滴加条件为每滴滴加间隔时间为2-3s。
优选的,所述反应时间为24-25h。
根据本发明优选的,步骤(5)中所述纳米fe3o4-sio2-pgma的制备,包括步骤:
将步骤(4)制备的纳米fe3o4-sio2-br均匀分散在甲基丙烯酸缩水甘油酯(gma)和引发剂异溴丁酸羟乙酯(hebib)的苯甲醚溶液中,液氮冷冻下脱气置换n2,加入催化剂cubr,分散均匀,再次进行液氮冷冻置换n2;加入催化剂配体五甲基二乙烯三胺(pmdeta),室温下解冻配位,于50-70℃聚合反应6-18h;经外加磁场分离、洗涤、干燥,得到纳米fe3o4-sio2-pgma;
优选的,所述异溴丁酸羟乙酯(hebib)与甲基丙烯酸缩水甘油酯(gma)的摩尔比为1:100-1:500,甲基丙烯酸缩水甘油酯与cubr的摩尔比为500:1-700:1,五甲基二乙烯三胺(pmdeta)与cubr的摩尔比为1:1-2:1,纳米fe3o4-sio2-br与甲基丙烯酸缩水甘油酯的质量比为1:50-1:150,所述甲基丙烯酸缩水甘油酯(gma)和苯甲醚的质量比为1:2-1:5。
优选的,所述聚合反应温度为60℃,反应时间为10-18h;所述洗涤条件为:使用四氢呋喃洗涤5次;所述干燥条件为真空干燥。
根据本发明优选的,步骤(6)中所述纳米fe3o4-sio2-pgma与赖氨酸(lysine)的质量比为1:5-1:20,na2co3与赖氨酸的摩尔比为2:1,纳米fe3o4-sio2-pgma与去离子水的质量比为1:500-1:800,去离子水与1,4-二氧六环的体积比为1:1。
根据本发明优选的,步骤(6)中所述反应温度为75℃,反应时间为24h。
根据本发明优选的,步骤(6)中所述惰性气体为氮气、氩气或氦气中的一种。
根据本发明优选的,步骤(6)中所述洗涤条件为:去离子水洗涤;所述干燥条件为:真空干燥。
本发明选择具有优良磁响应性能和较大比表面积的纳米fe3o4材料作为基材,首先,利用正硅酸四乙酯水解在纳米四氧化三铁表面原位包覆sio2层,利用3-氨丙基三乙氧基硅烷偶联剂(kh550)引入氨基以制备fe3o4-sio2-nh2,然后再与2-溴异丁酰溴通过亲和取代反应进行溴代改性制备纳米fe3o4-sio2-br,然后通过si-atrp(表面引发原子转移自由基聚合)活性聚合在纳米fe3o4-sio2-br表面接枝高密度的pgma聚合物刷,最后利用两性的赖氨酸功能性基团对聚合物刷中的环氧基团进行开环改性制备了高选择性磁性染料吸附剂。本发明的吸附剂材料表面聚合物刷的电荷随环境的ph变化呈现出不同的状态,因而能够对污水中的阴/阳离子型有机染料进行选择性的吸附脱除。此外,本发明的吸附剂能够在外界磁场作用下进行快速定向分离,有效避免了二次污染的发生。该种选择性高效磁性纳米有机染料吸附剂在有机染料污水治理领域的应用前景非常广阔。
本发明的有益效果如下:
1、本发明以磁性纳米四氧化三铁为基材,比表面积较大,显著提高了吸附容量;并且,具有良好的磁响应特点,能够在外界磁场作用下进行快速定向分离,简单易行,有效避免了二次污染的发生,可循环使用大幅降低生产成本。
2、本发明在四氧化三铁表面包覆二氧化硅能够有效抑制fe3o4内核的氧化进程,同时也提高了吸附剂在强酸或强碱环境下的稳定性,提高循环使用寿命;并且二氧化硅表面含有活性基团,易于改性。
3、本发明进行纳米fe3o4-sio2的表面氨基改性和纳米fe3o4-sio2-nh2溴代改性,保证了pgma聚合物刷能够以共价键的形式锚定在基材的表面,从而提高了吸附剂结构的稳定性。
4、本发明利用atrp活性聚合手段在纳米四氧化三铁表面接枝高密度pgma聚合物刷,有效抑制了纳米颗粒自身的团聚作用,同时引入了数量众多的活性环氧基团,进而利用两性的赖氨酸功能性基团通过开环反应引入丰富的吸附位点,有利于提高吸附容量。
5、本发明的吸附剂易通过磁场快速回收,表面接枝的经赖氨酸改性的高密度pgma聚合物刷为吸附剂带来了较高的吸附容量,两性赖氨酸基团可通过静电相互作用快速完成吸附过程,能够对污水中的阴/阳离子型有机染料进行选择性的高效吸附脱除;本发明选用特定的赖氨酸进行改性,能够实现在一个特定ph条件下只能吸附一种染料而对其它染料无吸附的特性,具有高度选择性。
6、本发明原料易得,工艺简单,所得吸附剂循环利用率高,实际应用价值高。
附图说明
图1是实施例1制备的高选择性磁性染料吸附剂的红外光谱图。
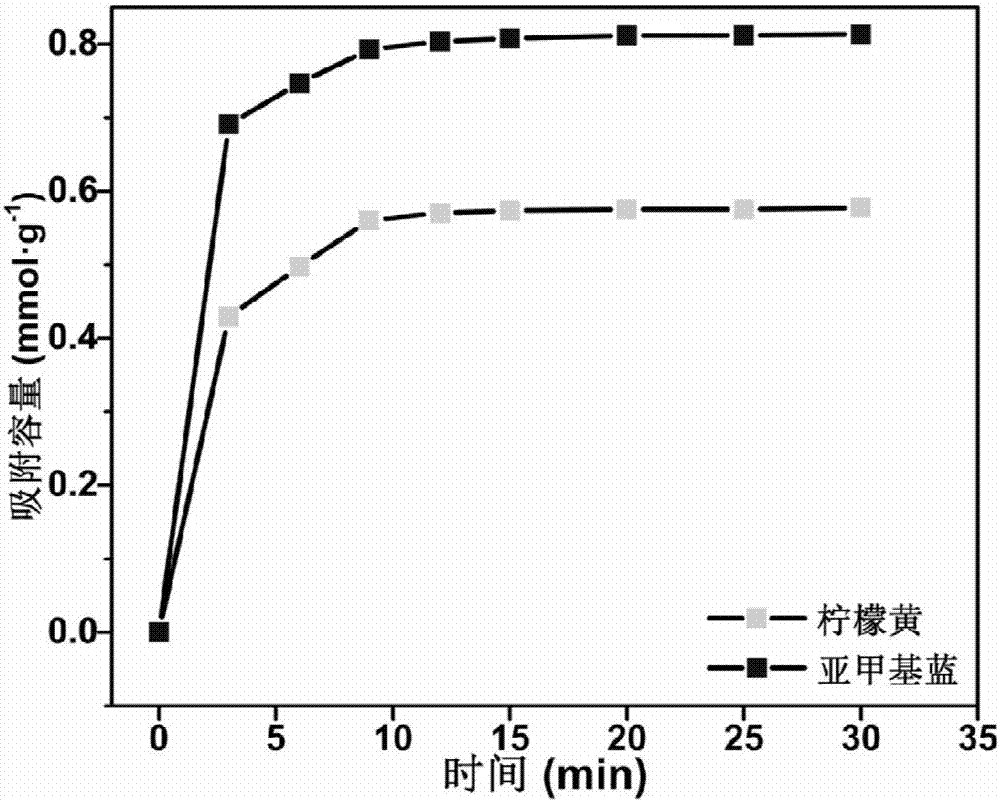
图2是实施例1制备的高选择性磁性染料吸附剂在25℃下对柠檬黄和亚甲基蓝有机染料的吸附动力学曲线。
具体实施方式
下面结合具体实施例对本发明做进一步的说明,但不限于此。
同时下述实施例中所述实验方法,如无特殊说明,均为常规方法;所述试剂和材料,如无特殊说明,均可从商业途径获得。
实施例1
一种高选择性磁性染料吸附剂的制备方法,步骤如下:
(1)磁性纳米fe3o4的制备
将2.7gfecl3·6h2o和7.2g无水naac超声均匀分散在100ml乙二醇中,室温下搅拌过夜,在高压反应釜中升温至200℃反应18h,外加磁场分离得到的固体物分散在无水乙醇中洗涤5次,然后室温真空干燥,得到磁性纳米fe3o4;
(2)纳米fe3o4-sio2的制备
将50mg步骤(1)得到的磁性纳米fe3o4超声分散在150ml无水乙醇中,加入5ml质量浓度为26%的氨水和3ml正硅酸四乙酯,升温至50℃并机械搅拌24h,外加磁场分离,无水乙醇和去离子水交替洗涤5次,室温真空干燥,得到纳米fe3o4-sio2;
(3)纳米fe3o4-sio2的表面氨基改性
将50mg步骤(2)制备的纳米fe3o4-sio2超声均匀分散在150ml无水乙醇中,加入1g3-氨丙基三乙氧基硅烷偶联剂(kh550),室温下搅拌2h,升温至85℃,并于n2氛围中回流,外加磁场分离,无水乙醇中洗涤5次,室温真空干燥,得到纳米fe3o4-sio2-nh2;
(4)纳米fe3o4-sio2-nh2溴代改性
将50mg步骤(3)制备的纳米fe3o4-sio2-nh2超声均匀分散在150ml二氯甲烷中,加入0.13g三乙胺,冰浴条件下向体系内缓慢滴加0.3g2-溴异丁酰溴,滴加速率为每滴间隔2-3s,室温下搅拌反应24h,外加磁场分离,去离子水和无水乙醇洗涤5次,室温真空干燥,得到纳米fe3o4-sio2-br;
(5)纳米fe3o4-sio2-pgma的制备
将50mg步骤(4)制备的纳米fe3o4-sio2-br超声均匀分散在溶有6ml甲基丙烯酸缩水甘油酯(gma)和20μl异溴丁酸羟乙酯(hebib)的20ml苯甲醚中,液氮冷冻置换n2两次后,将15mgcubr均匀分散在体系中,再次进行冷冻置换n2;加入25μl五甲基二乙烯三胺(pmdeta),室温下解冻配位,体系升温至60℃聚合反应12h;外加磁场分离,使用四氢呋喃洗涤5次,真空干燥,得到纳米fe3o4-sio2-pgma;
(6)纳米fe3o4-sio2-pgma的lysine改性
将50mg步骤(5)制备的纳米fe3o4-sio2-pgma分散于25ml去离子水和25ml1,4-二氧六环中,加入0.7gna2co3和0.5g赖氨酸(lysine),n2氛围下室温搅拌2h,升温至75℃搅拌反应24h,外加磁场分离,去离子水洗涤5次,真空干燥,即得。本实施例吸附剂的接枝率为86.90%。
图1是本实施例制备的吸附剂的红外光谱图,由图可知,本实施例制备的吸附剂具有式i所示结构。
吸附剂的有机染料吸附容量按照如下方法测定:准确称量一定质量(m)的吸附剂,加入至一系列同体积(v)不同浓度(c0)的ph=4的柠檬黄或ph=10的亚甲基蓝的有机染料溶液中。在25℃下于恒温震荡摇床中充分吸附,作用完毕后,利用外加磁场进行分离。利用紫外可见分光光度计(uv-vis)测定溶液中有机染料的平衡浓度(ce)。平衡吸附容量(qe)按照如下公式进行计算得到:
qe=(c0-ce)v/m
图2是本实施例制备的吸附剂对柠檬黄(质量浓度为1000mg·l-1)和亚甲基蓝(质量浓度为1000mg·l-1)有机染料的吸附动力学曲线。由图2可知,本实施例制备的吸附剂在25℃下,ph为4的条件下,对柠檬黄的吸附容量为0.567mmol·g-1;本实施例制备的吸附剂在25℃下,ph为10的条件下,对亚甲基蓝的吸附容量为0.813mmol·g-1。
本实施例所制备的吸附剂在柠檬黄有机染料中吸附饱和后在浓度为20mmol/l的nah2po4-naoh缓冲溶液中进行解吸附,然后再于ph=4的环境中对柠檬黄(质量浓度为1000mg·l-1)进行吸附,对柠檬黄的吸附容量为0.528mmol·g-1;本实施例所制备的吸附剂在亚甲基蓝有机染料中吸附饱和后在浓度为20mmol/l的naac-hac缓冲溶液中进行解吸附,然后再于ph=10的环境中对亚甲基蓝(质量浓度为1000mg·l-1)进行吸附,吸附容量为0.712mmol·g-1。
由上述可知,本发明的吸附剂可对污水中的阴/阳离子型有机染料进行选择性的高效吸附脱除,吸附容量大,吸附速率快,并且能够在外加磁场作用下实现循环利用和对有机染料的选择性回收,应用成本较低。
实施例2
一种高选择性磁性染料吸附剂的制备方法,如实施例1所述,所不同的是步骤(2)中所用正硅酸四乙酯的量为1ml,制备的吸附剂接枝率为51.60%。
按照实施例1的方法测试吸附剂对有机染料的吸附容量。本实施例制备的吸附剂在ph=4的环境中对柠檬黄(质量浓度为1000mg·l-1)的吸附容量为0.357mmol·g-1;在ph=10的环境中对亚甲基蓝(质量浓度为1000mg·l-1)的吸附容量为0.511mmol·g-1。
实施例3
一种高选择性磁性染料吸附剂的制备方法,如实施例1所述,所不同的是步骤(2)中所用正硅酸四乙酯的量为5ml,制备的吸附剂接枝率为88.60%。
按照实施例1的方法测试吸附剂对有机染料的吸附容量。本实施例制备的吸附剂在ph=4的环境中对柠檬黄(质量浓度为1000mg·l-1)的吸附容量为0.601mmol·g-1;在ph=10的环境中对亚甲基蓝(质量浓度为1000mg·l-1)的吸附容量为0.826mmol·g-1。
实施例4
一种高选择性磁性染料吸附剂的制备方法,如实施例1所述,所不同的是步骤(2)中反应温度为25℃,制备的吸附剂接枝率为57.40%。
按照实施例1的方法测试吸附剂对有机染料的吸附容量。本实施例制备的吸附剂在ph=4的环境中对柠檬黄(质量浓度为1000mg·l-1)的吸附容量为0.397mmol·g-1;在ph=10的环境中对亚甲基蓝(质量浓度为1000mg·l-1)的吸附容量为0.604mmol·g-1。
实施例5
一种高选择性磁性染料吸附剂的制备方法,如实施例1所述,所不同的是步骤(3)中反应温度为70℃,制备的吸附剂接枝率为64.20%。
按照实施例1的方法测试吸附剂对有机染料的吸附容量。本实施例制备的吸附剂在ph=4的环境中对柠檬黄(质量浓度为1000mg·l-1)的吸附容量为0.424mmol·g-1;在ph=10的环境中对亚甲基蓝(质量浓度为1000mg·l-1)的吸附容量为0.554mmol·g-1。
实施例6
一种高选择性磁性染料吸附剂的制备方法,如实施例1所述,所不同的是步骤(4)中室温反应时间为6h,制备的吸附剂接枝率为74.51%。
按照实施例1的方法测试吸附剂对有机染料的吸附容量。本实施例制备的吸附剂在ph=4的环境中对柠檬黄(质量浓度为1000mg·l-1)的吸附容量为0.447mmol·g-1;在ph=10的环境中对亚甲基蓝(质量浓度为1000mg·l-1)的吸附容量为0.589mmol·g-1。
实施例7
一种高选择性磁性染料吸附剂的制备方法,如实施例1所述,所不同的是步骤(5)中所用异溴丁酸羟乙酯(hebib)的加入量为40μl,制备的吸附剂接枝率为78.60%。
按照实施例1的方法测试吸附剂对有机染料的吸附容量。本实施例制备的吸附剂在ph=4的环境中对柠檬黄(质量浓度为1000mg·l-1)的吸附容量为0.497mmol·g-1;在ph=10的环境中对亚甲基蓝(质量浓度为1000mg·l-1)的吸附容量为0.642mmol·g-1。
实施例8
一种高选择性磁性染料吸附剂的制备方法,如实施例1所述,所不同的是步骤(5)中聚合所用时间为6h,制备的吸附剂接枝率为73.20%。
按照实施例1的方法测试吸附剂对有机染料的吸附容量。本实施例制备的吸附剂在ph=4的环境中对柠檬黄(质量浓度为1000mg·l-1)的吸附容量为0.414mmol·g-1;在ph=10的环境中对亚甲基蓝(质量浓度为1000mg·l-1)的吸附容量为0.537mmol·g-1。
实施例9
一种高选择性磁性染料吸附剂的制备方法,如实施例1所述,所不同的是步骤(5)中聚合所用时间为18h,制备的吸附剂接枝率为89.20%。
按照实施例1的方法测试吸附剂对有机染料的吸附容量。本实施例制备的吸附剂在ph=4的环境中对柠檬黄(质量浓度为1000mg·l-1)的吸附容量为0.608mmol·g-1;在ph=10的环境中对亚甲基蓝(质量浓度为1000mg·l-1)的吸附容量为0.811mmol·g-1。
实施例10
一种高选择性磁性染料吸附剂的制备方法,如实施例1所述,所不同的是步骤(6)中反应温度为50℃,制备的吸附剂接枝率为84.10%。
按照实施例1的方法测试吸附剂对有机染料的吸附容量。本实施例制备的吸附剂在ph=4的环境中对柠檬黄(质量浓度为1000mg·l-1)的吸附容量为0.346mmol·g-1;在ph=10的环境中对亚甲基蓝(质量浓度为1000mg·l-1)的吸附容量为0.484mmol·g-1。
实施例11
一种高选择性磁性染料吸附剂的制备方法,如实施例1所述,所不同的是步骤(6)中反应温度为室温下反应,制备的吸附剂接枝率为83.60%。
按照实施例1的方法测试吸附剂对有机染料的吸附容量。本实施例制备的吸附剂在ph=4的环境中对柠檬黄(质量浓度为1000mg·l-1)的吸附容量为0.257mmol·g-1;在ph=10的环境中对亚甲基蓝(质量浓度为1000mg·l-1)的吸附容量为0.366mmol·g-1。
实施例12
一种高选择性磁性染料吸附剂的制备方法,如实施例1所述,所不同的是步骤(6)中所用反应时间为6h,制备的吸附剂接枝率为85.50%。
按照实施例1的方法测试吸附剂对有机染料的吸附容量。本实施例制备的吸附剂在ph=4的环境中对柠檬黄(质量浓度为1000mg·l-1)的吸附容量为0.474mmol·g-1;在ph=10的环境中对亚甲基蓝(质量浓度为1000mg·l-1)的吸附容量为0.608mmol·g-1。
- 还没有人留言评论。精彩留言会获得点赞!