复合纳米光催化剂及制备方法与流程
本发明属于无机复合纳米催化材料制备技术领域,具体涉及一种cop纳米颗粒修饰c3n4纳米粉体的复合纳米催化剂及制备方法。
背景技术
氢能,作为二次能源被认为是最理想的绿色能源之一。目前成熟的制氢技术中,化石燃料制氢不能根本解决能源短缺和环境污染问题,电解水制氢成本较高,高温热解水制氢能量转化效率较低,都难以发展为能源氢的生产技术。因此,开发清洁、高效、低成本的制氢技术,具有重大的社会和经济意义。
c3n4具有稳定性好、无毒、成本低、来源广泛以及光电性能优异等优点,是目前研究最广泛,也是最具有开发前景的光解水催化剂。然而,c3n4的禁带宽度(约3.2~3.4ev)较大,只能对波长小于387nm的太阳光进行有效吸收,这部分只占太阳光谱总能量不到10%,而对其余占太阳光总能量>90%的可见和红外辐射无法利用;同时,c3n4纳米晶光催化剂的光致电荷分离态(电子-空穴对)易于复合,导致光量子效率降低,通常不会超过10%。这两点使得传统c3n4光催化技术的总体效率非常有限,极大的限制了其推广应用。
窄禁带半导体修饰技术可以有效扩展半导体纳米晶的光谱响应范围,提高光致电荷分离的量子产率,从而优化其光催化效率。cop是一类性能优异的窄带半导体,具有下列特点:1)较宽的光谱响应范围,从而具有很好的吸光能力;2)较小的电子重组能和较强的给电子能力,从而可以提高纳米晶界面的电荷分离能力,提高光能利用效率;3)较高的光稳定性、化学稳定性和热稳定性,保证较长的使用寿命和复杂环境适应性。
上述特点表明cop特别适合于作为c3n4纳米晶的修饰剂,用于复合光催化功能材料的制备,是一种有效的提高光催化剂的重要方法。
技术实现要素:
本发明的目的是提供一种c3n4/cop复合纳米光催化剂及其制备方法。本发明通过窄带半导体cop修饰的方式可以在一定程度上解决目前c3n4存在的光电-空穴复合率高、光量子效率低等问题,提高c3n4的可见光响应性和光催化活性;同时该复合光催化剂的制备方法简单,工艺条件易调控、成本低、易于工业生产和推广应用。
一种c3n4/cop复合纳米光催化剂,该复合纳米光催化剂的组分含有c3n4纳米粉体和cop纳米颗粒,所述cop与c3n4的质量比为1-5:100,具有复合纳米结构。
所述cop负载在c3n4表面,cop在c3n4表面的负载量为1%-5%;
一种c3n4/cop复合纳米光催化剂的制备方法,包括如下步骤:
(1)制备c3n4纳米粉体;
首先,g-c3n4纳米片采用热解的方法制取,取10-15g尿素置于坩埚中,改好盖子,以每分钟10℃的速度缓慢升温至500℃,恒温保持5小时。将反应后得到的固体冷却至室温,研磨后收集起来,得到c3n4纳米粉体;
(2)配制敏化液:称取二水氯化亚锡0.5-1g,超声溶解在2.5-5ml12mol/l盐酸中加蒸馏水定容至0.5-1l配成敏化液;
(3)配制活化液:称取氯化钯0.05-0.1g,超声溶解在0.5-1ml12mol/l盐酸中加蒸馏水定容至1l配成活化液。
(4)配制镀液:六水氯化钴0.5945-1.189g,次亚磷酸钠2.12-4.24g,甘氨酸1.126-2.252g,加至蒸馏水80ml,待完全溶解,加入氢氧化钠溶液,调节溶液ph值为11,并转移到100ml容量瓶后作为镀液待用;
(5)制备cop负载c3n4纳米复合材料,将步骤(1)得到c3n4纳米粉体和步骤(4)得到的镀液混合,超声,搅拌,均匀后,水浴加热60℃,反应3-5小时;将得到的产物用蒸馏水和乙醇交替离心洗涤;然后在真空环境下,温度为60-70℃温度范围内干燥3-5小时,进行干燥,以保证将c3n4/cop样品得到了充分干燥。
作为优选的,一种c3n4/cop复合纳米光催化剂的制备方法,包括如下步骤:
(1)制备c3n4纳米粉体;
首先,g-c3n4纳米片采用热解的方法制取,取10g尿素置于坩埚中,以每分钟10℃的速度缓慢升温至500℃,恒温保持5小时。将反应后得到的固体冷却至室温,研磨后收集起来,得到c3n4纳米粉体;
(2)配制敏化液:称取二水氯化亚锡1g,超声溶解在5ml12mol/l盐酸中加蒸馏水定容至1l配成敏化液;
(3)配制活化液:称取氯化钯0.1g,超声溶解在1ml12mol/l盐酸中加蒸馏水定容至1l配成活化液。
(4)配制镀液:六水氯化钴1.189g,次亚磷酸钠4.24g,甘氨酸2.252g,加至蒸馏水80ml,待完全溶解,加入氢氧化钠溶液,调节溶液ph值为11,并转移到100ml容量瓶后作为镀液待用;
(5)制备cop负载c3n4纳米复合材料,将步骤(1)得到c3n4纳米粉体和步骤(4)得到的镀液混合,超声,搅拌,均匀后,水浴加热60℃,反应3小时;将得到的产物用蒸馏水和乙醇交替离心洗涤;然后在真空环境下,温度为60-70℃温度范围内干燥3小时,进行干燥,以保证将c3n4/cop样品得到了充分干燥。
发明的有益效果:
采用热解法制备的一定量的c3n4纳米粉体,再通过采用化学镀的方法,制备cop负载c3n4纳米复合材料;利用漫反射和吸收光谱分析发现复合材料在可见和近红外区的光谱响应比单纯c3n4纳米粉体得到了显著加强,制备过程简洁,得到的复合催化材料性能稳定,性价比高,适宜于大规模生产,用于光催化分解水产氢。
附图说明
下面结合附图对本发明作进一步的说明。
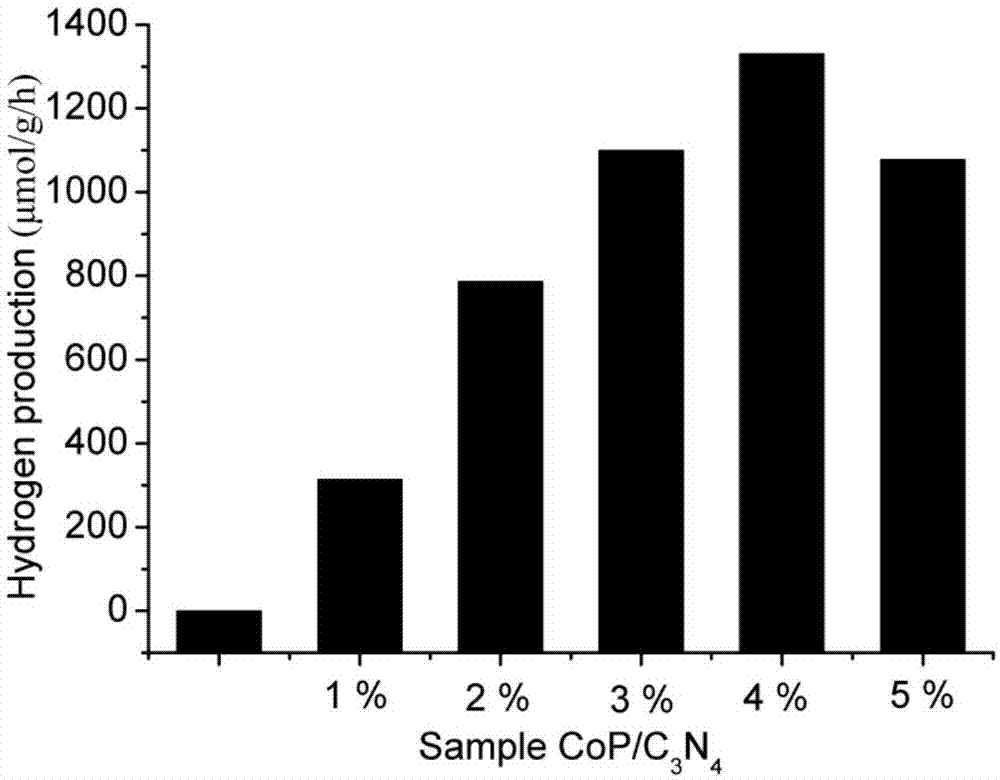
图1是实施例1所制备的c3n4和c3n4/cop样品对光催化分解水产氢的活性图;
图2是实施例1所制备的c3n4和c3n4/cop复合材料的xrd谱图;
图3是紫外-可见漫反射(uv-drs)光谱;
图4是c3n4/cop纳米复合光催化剂的xps谱图。
具体实施方式
实施例1
采用热解法制备的一定量的c3n4纳米粉体,再通过采用化学镀的方法,制备cop不同负载量的c3n4/cop纳米复合材料;首先,g-c3n4纳米片采用热解的方法制取,取10g尿素置于坩埚中,盖好盖子,以每分钟10℃的速度缓慢升温至500℃,恒温保持5小时;将反应后得到的固体冷却至室温,研磨后收集起来,得到c3n4纳米粉体。
称取二水氯化亚锡1g,超声溶解在5ml盐酸中加蒸馏水定容至1l配成敏化液。称取氯化钯0.1g,超声溶解在1ml盐酸中加蒸馏水定容至1l配成活化液。六水氯化钴1.189g,次亚磷酸钠4.24g,甘氨酸2.252g,加至蒸馏水80ml,待完全溶解,加入氢氧化钠溶液,调节溶液ph值为11,并转移到100ml容量瓶后作为镀液待用。
接下来,制备4.0wt%cop负载c3n4纳米复合材料。将1.0g的c3n4纳米粉体和4.0ml的镀液混合,超声,搅拌,均匀后,水浴加热60℃,反应5小时。将得到的产物用蒸馏水和乙醇交替离心洗涤;然后在真空环境下,温度为100℃温度范围内干燥5小时,进行干燥,以保证将c3n4/cop(4wt%cop)样品得到了充分干燥。
本发明c3n4/cop(4wt%cop)光催化剂的光催化活性是通过模拟太阳光照下光催化分解水产氢进行测定的,具体如下:
光催化反应过程采用自制的100ml的三口烧瓶作为光催化反应体系,功率为300w的氙灯作为反应光源,测试过程中玻璃反应器距氙灯水平距离为20cm。实验中采用三乙醇胺作为反应的牺牲剂,其中三乙醇胺的体积分数为15%。具体实验过程中,将50mgc3n4/cop(4wt%cop)样品加入68ml水和12ml牺牲剂中,将玻璃反应器常温超声处理5min,在进行光催化反应之前向装置中通氮气30min以排尽装置中的空气来保证反应器内的厌氧环境,排除装置中的分子氧对反应结果造成的影响。待装置在氙灯下照射1小时后,用微量注射器取0.4ml反应后的气体在气相色谱(gc-14c,shimadzu,日本,tcd,n2作为载气,
图2为实施例1所制备c3n4和c3n4/cop复合纳米光催化剂材料的xrd谱图比较,可以看出4wt%cop修饰后的c3n4复合纳米材料仍然保持着c3n4的晶体结构。图3给出c3n4纳米粉体和实施例1所制备的c3n4/cop(4wt%cop)纳米光催化剂复合材料的紫外-可见吸收光谱比较,从图3中可以清楚地看出,c3n4/cop复合材料在400nm区域的吸光能力很强,而未加修饰的c3n4在该区域没有吸收。图4是实施例1所制备的c3n4/cop(4wt%cop)纳米复合光催化剂的xps谱图,表示cop纳米颗粒成功的镀到了c3n4纳米粉体的表面。
实施例2
采用热解法制备的一定量的c3n4纳米粉体,再通过采用化学镀的方法,制备cop不同负载量的c3n4/cop纳米复合材料;首先,g-c3n4纳米片采用热解的方法制取,取10g尿素置于坩埚中,改好盖子,以每分钟10℃的速度缓慢升温至500℃,恒温保持5小时。将反应后得到的固体冷却至室温,研磨后收集起来,得到c3n4纳米粉体。
称取二水氯化亚锡1g,超声溶解在5ml盐酸中加蒸馏水定容至1l配成敏化液。称取氯化钯0.1g,超声溶解在1ml盐酸中加蒸馏水定容至1l配成活化液。六水氯化钴1.189g,次亚磷酸钠4.24g,甘氨酸2.252g,加至蒸馏水80ml,待完全溶解,加入氢氧化钠溶液,调节溶液ph值为11,并转移到100ml容量瓶后作为镀液待用。
接下来,制备1.0wt%cop负载c3n4纳米复合材料,将1.0g的c3n4纳米粉体和1.0ml的镀液混合,超声,搅拌,均匀后,水浴加热60℃,反应5小时。将得到的产物用蒸馏水和乙醇交替离心洗涤;然后在真空环境下,温度为100℃温度范围内干燥5小时,进行干燥,以保证将c3n4/cop(1wt%cop)样品得到了充分干燥。
本发明c3n4/cop(1wt%cop)光催化剂的光催化活性是通过模拟太阳光照下光催化分解水产氢进行测定的,具体如下:
光催化反应过程采用自制的100ml的三口烧瓶作为光催化反应体系,功率为300w的氙灯作为反应光源,测试过程中玻璃反应器距氙灯水平距离为20cm。实验中采用三乙醇胺作为反应的牺牲剂,其中三乙醇胺的体积分数为15%。具体实验过程中,将50mgc3n4/cop(1wt%cop)样品加入68ml水和12ml牺牲剂中,将玻璃反应器常温超声处理5min,在进行光催化反应之前向装置中通氮气30min以排尽装置中的空气。待装置在氙灯下照射1小时后,用微量注射器取0.4ml反应后的气体在气相色谱上进行分析检测。光催化反应全部都是在不断的搅拌的条件下进行。如图1是所制备的c3n4和c3n4/cop(1wt%cop)样品对光催化分解水制氢的活性图;以本方法制备的c3n4/cop(1wt%cop)复合光催化剂,在模拟太阳光下,相比纯的c3n4纳米粉体,其光催化活性有了一定的提高。
实施例3
采用热解法制备的一定量的c3n4纳米粉体,再通过采用化学镀的方法,制备cop不同负载量的c3n4/cop纳米复合材料;首先,g-c3n4纳米片采用热解的方法制取,取10g尿素置于坩埚中,改好盖子,以每分钟10℃的速度缓慢升温至500℃,恒温保持5小时。将反应后得到的固体冷却至室温,研磨后收集起来,得到c3n4纳米粉体。
称取二水氯化亚锡1g,超声溶解在5ml盐酸中加蒸馏水定容至1l配成敏化液。称取氯化钯0.1g,超声溶解在1ml盐酸中加蒸馏水定容至1l配成活化液。六水氯化钴1.189g,次亚磷酸钠4.24g,甘氨酸2.252g,加至蒸馏水80ml,待完全溶解,加入氢氧化钠溶液,调节溶液ph值为11,并转移到100ml容量瓶后作为镀液待用。
接下来,制备5.0wt%cop负载c3n4纳米复合材料。将1.0g的c3n4纳米粉体和5.0ml的镀液混合,超声,搅拌,均匀后,水浴加热60℃,反应5小时。将得到的产物用蒸馏水和乙醇交替离心洗涤;然后在真空环境下,温度为100℃温度范围内干燥5小时,进行干燥,以保证将c3n4/cop(5wt%cop)样品得到了充分干燥。
本发明c3n4/cop(5wt%cop)光催化剂的光催化活性是通过模拟太阳光照下光催化分解水产氢进行测定的,具体如下:
光催化反应过程采用自制的100ml的三口烧瓶作为光催化反应体系,功率为300w的氙灯作为反应光源,测试过程中玻璃反应器距氙灯水平距离为20cm。实验中采用三乙醇胺作为反应的牺牲剂,其中三乙醇胺的体积分数为15%。具体实验过程中,将50mgc3n4/cop(5wt%cop)样品加入68ml水和12ml牺牲剂中,将玻璃反应器常温超声处理5min,在进行光催化反应之前向装置中通氮气30min以排尽装置中的空气。待装置在氙灯下照射1小时后,用微量注射器取0.4ml反应后的气体在气相色谱上进行分析检测。光催化反应全部都是在不断的搅拌的条件下进行。如图1是所制备的c3n4和c3n4/cop(5wt%cop)样品对光催化分解水制氢的活性图;以本方法制备的c3n4/cop(5wt%cop)复合光催化剂,在模拟太阳光下,相比纯的c3n4纳米粉体,其光催化活性有了一定的提高,但是其光催化活性略低于c3n4/cop(4wt%cop)。
- 还没有人留言评论。精彩留言会获得点赞!
- 一种四角型矾酸铋/石墨烯复合光催化剂的制备方法与制造工艺
- 硅藻土/纳米TiO2光催化涂料添加剂的稳定化方法与制造工艺
- 一种具有光催化特性的核壳结构杂化溶胶及其制备与应用的制造方法与工艺
- 一种双粒径分布的纳米二氧化钛光催化剂及其制备方法与制造工艺
- 一种用于排气处理的纳米催化器的制造方法
- 以玻璃纤维织物为基底的二氧化钛纳米线阵列光催化剂的制作方法
- 一种制备纳米催化材料的工艺装置的制造方法
- 一种MIL?53(Fe)/g?C<sub>3</sub>N<sub>4</sub>纳米片复合光催化材料的制备方法
- 高效Ag/AgCl空心八面体可见光光催化剂的制备方法
- 一种制备CdS/CoWO<sub>4</sub>异质结复合光催化剂的方法