一种氨硼烷水解析氢用催化剂及其制备方法
1.本发明属于氨硼烷水解析氢技术领域,具体涉及一种氨硼烷水解析氢用催化剂及其制备方法。
背景技术:
2.氢气,作为一种清洁能源的载体,具有能量密度高、燃烧后无污染等优点,是最具可持续发展潜力的清洁能源载体。nh3bh3作为固态储氢材料,具有高氢含量(19.6wt%)、低分子量(30.87g/mol)、常温下稳定、无毒等特点在引起了研究者的广泛关注。贵金属催化剂具有优异的催化活性,但是价格昂贵、地球含量低限制了贵金属的广泛应用。研究开发低成本、高活性的非贵金属催化剂是氨硼烷水解析氢的关键科学问题之一,也是目前该领域的研究热点和重点。
3.co/ni作为过渡金属具有构建活性位点以提高催化性能的潜力,但是在水解产氢反应中产生的低价b元素具有很强的还原性,导致催化活性降低。过渡金属磷化物作为一种活性材料,由于其丰富的活性位点、独特的理化性质和可调节的组分结构等优点,在提高催化剂的活性方面具有广泛的应用。然而,在磷化物的合成过程中,磷原子取代引起的晶格应力会导致金属磷化物团聚,导致催化活性降低。碳载体的存在可以分散纳米颗粒对进一步提高活性组分的催化活性和稳定性具有重要意义,选用合适的催化剂载体并加强金属与载体之间的电子转移是提升非贵金属基催化剂活性的关键。另外,在过渡金属磷化物中引入异质原子o元素,可以对反应分子的反应性能进行协同调控,促进催化性能。但是氨硼烷水解产氢的活性有待进一步提升。
技术实现要素:
4.针对上述现有技术的缺陷与不足,本发明的目的在于提供一种通过可控氧化和表面磷化制备氨硼烷水解析氢用催化剂o-(cop/co2p)@sc及其制备方法,解决如下几处问题:
①
贵金属基催化剂在氨硼烷水解产氢中价格昂贵、资源受限;
②
非贵金属在制备的过程中容易聚集团聚;
③
非贵金属催化剂在氨硼烷水解产氢中活性低。
5.为实现上述目的,本发明氨硼烷水解析氢用催化剂的技术方案如下:
6.一种氨硼烷水解析氢用催化剂,所述催化剂的分子式为o-(cop/co2p)@sc,结构为o修饰的cop和co2p异质结构负载到碳载体上。
7.本发明提供一种氨硼烷水解析氢用催化剂的制备方法,采用如下技术方案,制备步骤如下:
8.s1:将0.75g六水硝酸钴加入到48ml的n,n-二甲基甲酰胺中,得到溶液ⅰ;
9.s2:将0.428g对苯二甲酸加入到12ml乙醇中,得到溶液ⅱ;
10.s3:将s1中制备的溶液ⅰ加入到s2中制备的溶液ⅱ中,得到溶液ⅲ;
11.s4:将s3中制备的溶液ⅲ在150℃温度下保温12h,冷却到室温,超声分散2h形成co-mof溶液;
12.s5:将4ml乙醇加入到16ml水中得到溶液ⅳ;
13.s6:将0.08ml硅酸四乙酯加入到s5中制备的溶液ⅳ中,得到溶液
ⅴ
;
14.s7:将s6中制备的溶液
ⅴ
加入到s4中制备的co-mof溶液中,搅拌8h,得到co-mof@sio2溶液;
15.s8:将54ml乙醇加入到26ml水中,得到溶液ⅵ;
16.s9:将0.17g间苯二酚和0.17ml乙二胺加入到s8中制备的溶液ⅵ中,搅拌30min,得到溶液ⅶ;
17.s10:将0.26ml甲醛加入到s9中制备的溶液ⅶ中,搅拌30min,得到间苯二酚-甲醛树脂溶液;
18.s11:将s7中制备的co-mof@sio2溶液滴加到s10中制备的间苯二酚-甲醛树脂溶液中,30℃下水浴搅拌48h,然后50℃烘干12h,得到co-mof@sio2@rf前驱体;
19.s12:将s11中制备的co-mof@sio2@rf前驱体在氩气氢气混合气氛下700℃保温2h,冷却至室温;
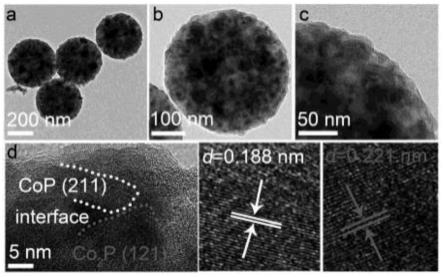
20.s13:将10ml乙醇加入到s12制备的煅烧材料中,30℃烘干12h,制得催化剂co@sio2@c(co@sc);
21.s14:将s13中制备的催化剂co@sio2@c(co@sc)进行可控氧化和表面磷化,得到o-(cop/co2p)@sc催化剂。
22.优选的,步骤s12中,所述的氩气氢气混合气氛中氩气与氢气的体积比为99∶1。
23.优选的,步骤s12中,以5℃/min的速率从室温升温至700℃;
24.优选的,步骤s14中,所述的可控氧化温度为200℃,时间为22h;
25.优选的,步骤s14中,表面磷化的磷源为nah2po2,s14中的得到的催化剂与nah2po2的质量比为1∶10,表面磷化的温度为300℃,升温速率为2℃/min,时间为120min。
26.有益技术效果
27.1.通过o修饰的异质结构cop和co2p的引入,为催化氨硼烷水解产氢提供了必不可少的活性物质,降低了氨硼烷和水的解离能垒,优化了氨硼烷水解产氢的解离路线,提升了氨硼烷水解产氢活性。
28.2.创新性的合成了o修饰的异质结构cop和co2p催化剂,碳载体的存在使得cop和co2p颗粒大小均一、分布均匀。由此进一步处理得到的cop和co2p纳米颗粒可用于高效催化氨硼烷水解产氢,拓宽了催化剂的应用领域。
29.3.成本低、性能好。采用非贵金属所制备的o-(cop/co2p)@sc催化剂具有极高的氨硼烷催化活性,tof值达到35min-1
,优于行业内的同类型催化剂的tof(8.4min-1
)。
附图说明
30.图1:o-(cop/co2p)@sc(实施例1)、coo
x
@sc(对比例4)和co@sc(对比例5)的x射线粉末衍射图;
31.图2:o-(cop/co2p)@sc(实施例1)的co 2p和o1s的xps谱图;
32.图3:o-(cop/co2p)@sc(实施例1)的tem和hrtem图;
33.图4:o-(cop/co2p)@sc(实施例1)、co-p@sc(对比例3)、coo
x
@sc(对比例4)和co@sc(对比例5)的催化活性图。
具体实施方式
34.为使本发明更加清楚、明确,以下对本发明进一步详细说明。应当理解,此处所描述的具体实施例仅用以解释本发明,并不用于限定本发明。
35.实施例1
36.一种氨硼烷水解析氢用催化剂,分子式为o-(cop/co2p)@sc,结构为o修饰的cop和co2p异质结构负载到碳载体上。制备方法如下:
37.s1:将0.75g六水硝酸钴加入到48ml的n,n-二甲基甲酰胺中,得到溶液ⅰ;
38.s2:将0.428g对苯二甲酸加入到12ml乙醇中,得到溶液ⅱ;
39.s3:将s1中制备的溶液ⅰ加入到s2中制备的溶液ⅱ中,得到溶液ⅲ;
40.s4:将s3中制备的溶液ⅲ在150℃温度下保温12h,冷却到室温,超声分散2h形成co-mof溶液;
41.s5:将4ml乙醇加入到16ml水中得到溶液ⅳ;
42.s6:将0.08ml硅酸四乙酯加入到s5中制备的溶液ⅳ中,得到溶液
ⅴ
;
43.s7:将s6中制备的溶液
ⅴ
加入到s4中制备的co-mof溶液中,搅拌8h,得到co-mof@sio2溶液;
44.s8:将54ml乙醇加入到26ml水中,得到溶液ⅵ;
45.s9:将0.17g间苯二酚和0.17ml乙二胺加入到s8中制备的溶液ⅵ中,搅拌30min,得到溶液ⅶ;
46.s10:将0.26ml甲醛加入到s9中制备的溶液ⅶ中,搅拌30min,得到间苯二酚-甲醛树脂溶液;
47.s11:将s7中制备的co-mof@sio2溶液滴加到s10中制备的间苯二酚-甲醛树脂溶液中,30℃下水浴搅拌48h,然后50℃烘干12h,得到co-mof@sio2@rf前驱体;
48.s12:将s11中制备的co-mof@sio2@rf前驱体在氩气氢气混合气氛下700℃保温2h,冷却至室温;
49.s13:将10ml乙醇加入到s12制备的煅烧材料中,30℃烘干12h,制得催化剂co@sio2@c(co@sc);
50.s14:将s13中制备的催化剂co@sio2@c(co@sc)进行可控氧化和表面磷化,得到o-(cop/co2p)@sc催化剂。
51.优选的,步骤s12中,所述的氩气氢气混合气氛中氩气与氢气的体积比为99∶1。
52.优选的,步骤s12中,以5℃/min的速率从室温升温至700℃;
53.优选的,步骤s14中,所述的可控氧化温度为200℃,时间为22h;
54.优选的,步骤s14中,表面磷化的磷源为nah2po2,s14中的得到的催化剂与nah2po2的质量比为1∶10,表面磷化的温度为300℃,升温速率为2℃/min,时间为120min。
55.对比例1
56.与实施例1的不同之处在于:步骤s12中,温度为600℃,其它均同实施例1。
57.对比例2
58.与实施例1的不同之处在于:步骤s14中,温度为800℃,其它均同实施例1。
59.对比例3
60.与实施例1的不同之处在于:步骤s14中,不经过可控氧化,其它均同实施例1。
61.所得目标产品为co-p@sc。
62.对比例4
63.与实施例1的不同之处在于:步骤s14中,不经过表面磷化,其它均同实施例1。
64.所得目标产品为coo
x
@sc。
65.对比例5
66.与实施例1的不同之处在于:步骤s14中,不经过可控氧化和表面磷化,其它均同实施例1。
67.所得目标产品为co@sc。
68.催化剂结构表征
69.图1为制备的催化剂o-(cop/co2p)@sc(实施例1)、coo
x
@sc(对比例2)和co@sc(对比例3)的x射线粉末衍射图。从图1中可以看出:所制备催化剂与标准图谱卡片co(jcpds card no.15-0806)、co3o4(jcpds card no.42-1467)、cop(jcpds card no.89-2747)co2p(jcpds card no.32-0306)相对应。这表明催化剂在处理的过程中通过可控氧化和表面磷化策略目标催化剂o-(cop/co2p)@sc成功合成。
70.图2为制备的催化剂o-(cop/co2p)@sc(实施例1)的co 2p和o1s的xps图谱。co 2p和o1s的xps图谱显示o-(cop/co2p)@sc催化剂成功制备,并且co向p转移了电子。
71.图3为制备的催化剂o-(cop/co2p)@sc(实施例1)的tem和hrtem表征。从图3中可以看出cop和co2p异质结构成功合成,这和xrd结果保持一致。
72.催化剂性能测试
73.将制备的o-(cop/co2p)@sc(实施例1)、co-p@sc(对比例3)、coo
x
@sc(对比例4)和co@sc(对比例5)分别作为催化剂用于氨硼烷水解制氢。将10mg催化剂、5ml水溶液超声形成均匀的混合液后放入25ml的圆底烧瓶中,将0.045mg的氨硼烷加入上述烧瓶中,水解产氢反应是在水浴30℃的条件下进行的,每5ml记录产生的氢气体积与所需时间。
74.图4为不同催化剂氨硼烷水解析出氢气的实验结果,从图4(a)中可以看出:同样的条件下o-(cop/co2p)@sc(实施例1)相比于其他材料催化的反应所需时间最短,或相应从图4(b)可以看出o-(cop/co2p)@sc的tof远远高于其他对比催化剂。最高活性为35min-1
。表明了o修饰的异质结构磷化物催化剂极大地提升了催化剂在氨硼烷水解中的催化活性。
75.实施例和对比例的结果见表1。
76.表1
77.项目tof(min-1
)实施例135对比例121.6对比例215对比例311.9对比例415.8对比例58.6
78.从对比例中可以看出:
①
改变前驱体锻烧温度,得到的催化剂活性差;
②
只经过表面磷化处理的催化剂具有比较低的tof值;
③
只经过表面氧化处理的催化剂具有比较低的tof值;
④
不经过表面磷化和表面氧化处理的催化剂具有最低的tof值。
79.以上述依据本发明的理想实施例为启示,通过上述的说明内容,相关工作人员完全可以在不偏离本项发明技术思想的范围内,进行多样的变更以及修改。本项发明的技术性范围并不局限于说明书上的内容,必须要根据权利要求范围来确定其技术性范围。凡根据本发明精神实质所作的等效变化或修饰,都应涵盖在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1