一种Z型光催化剂及其制备方法和应用
一种z型光催化剂及其制备方法和应用
技术领域
1.本发明涉及光催化剂技术领域,尤其是涉及一种z型光催化剂及其制备方法和应用。
背景技术:
2.能源短缺和环境污染是人类社会发展面临的两大重要问题。因此新型清洁能源的开发利用和环境中污染物的防护治理工作成为了当前的研究焦点。在众多新型清洁能源中,氢能凭借着其较高的热值和极佳的环境效应,成为最具发展潜力的研究对象。氢能源的开发是一项热点课题,其中光催化技术利用太阳能辐射将水分解为氢气和氧气,是获取氢能源的理想途径。同时,光催化技术也被广泛用于降解液相和气相中的有机污染物,其利用光能实现有机污染物的彻底矿化,从而消除对环境的危害,是一项高效且环保的净化技术。可见,发展光催化技术是应对能源短缺和环境污染两大问题的有效举措。
3.选择合适的催化剂是光催化技术应用的关键。铋系半导体材料是近年来广受关注的一类新型光催化剂,其中bivo4半导体材料因具有无毒、地球丰度高、在水溶液中稳定性高、以及良好的吸光性能和较小的带隙(约2.40ev),被认为是最具发展前景的半导体催化材料之一。虽然bivo4是一种优秀的半导体材料,但是未经改性的单一bivo4光催化活性仍然低下,无法达到实际应用的要求,主要原因是其光生电子-空穴的分离效率低且复合速率快。通过构建异质结可提高半导体催化剂效果,进而目前提出了一些bivo4基异质结光催化剂,但现有的bivo4基异质结光催化剂一般仅对有机污染物具有氧化活性,无法通过光催化分解水产生h2,作用有限,对新型清洁能源的开发利用不能产生作用,由此限制了其应用范围。
技术实现要素:
4.本发明旨在至少解决现有技术中存在的技术问题之一。为此,本发明提出一种z型光催化剂及其制备方法和应用。
5.本发明的第一方面,提出了一种z型光催化剂的制备方法,包括bivo4/biocl p-n型异质结光催化剂、金属层和cds纳米颗粒,所述bivo4/biocl p-n型异质结光催化剂包括biocl纳米片和负载于所述biocl纳米片表面的bivo4纳米颗粒;所述金属层负载于所述bivo4纳米颗粒的表面,所述金属层的材质为au;所述cds纳米颗粒负载于所述金属层的表面。
6.根据本发明实施例z型光催化剂,至少具有以下有益效果:该z型光催化剂包括含biocl纳米片和负载于biocl纳米片表面的bivo4纳米颗粒的bivo4/biocl p-n型异质结光催化剂、负载在bivo4纳米颗粒表面的金金属层,以及负载于金金属层表面的cds纳米颗粒。其中,bivo4/biocl p-n型异质结光催化剂具有零维-二维分级纳米结构,具体为零维纳米颗粒和二维纳米片组装而成的分级复合结构,该分级复合结构可以防止低维纳米颗粒的团聚,保证纳米材料自身结构和形貌不被破坏,并且其含有较大的比表面积,具有较高的光生
载流子分离效率,对有机污染物具有优异的氧化活性;cds具有优异的光解水产氢性能,但单一的cds催化剂具有光腐蚀严重的问题,而以上以bivo4/biocl p-n型异质结光催化剂作为氧化端,cds纳米颗粒作为还原端,并以金金属层作为cds纳米颗粒定向负载的锚定位点,同时充当还原端催化剂和氧化端催化剂之间的电子中介体,依据载流子迁移路径,在bivo4/biocl p-n型异质结光催化剂中的bivo4端通过金金属层定向负载cds纳米颗粒,构建具有载流子双分离通道的z型光催化剂,所构建z型光催化剂体系可提供载流子分离的双通道,实现有效的光生电子和空穴分离,并且可有效抑制单一的cds催化剂光腐蚀严重的问题,充分发挥氧化段和还原端催化剂的氧化还原活性,使得产品z型光催化剂同时具备氧化降解有机物和光催化产氢能力;并且,通过将还原端催化剂选择性负载在氧化端催化剂的电子富集区域bivo4纳米颗粒上,可有效提升光生电子和空穴的分离效率,进而利用z型体系对光生载流子的有效分离,可进一步提升催化剂的光催化活性。由上,该z型光催化剂同时具备氧化降解有机物和光催化产氢能力,催化活性高,可应用于含有机物废水的处理,可实现光催化剂降解有机物同步制氢。
7.在本发明的一些实施方式中,所述bivo4纳米颗粒的粒径为80~100nm;优选地,所述biocl纳米片的平面尺寸为0.5~1μm,厚度为20~30nm。
8.在本发明的一些实施方式中,所述z型光催化剂中所述金属层的质量占比为1~2.5wt%,优选为2wt%。所述金属层由金纳米颗粒负载于bivo4纳米颗粒的表面构成。
9.在本发明的一些实施方式中,所述z型光催化剂中所述cds纳米颗粒的质量占比为5~20wt%,优选为10wt%。
10.在本发明的一些实施方式中,所述金属层的厚度为15~25nm,所述cds纳米颗粒的粒径为10~20nm。
11.本发明的第二方面,提出了一种以上任一种z型光催化剂的制备方法,包括以下步骤:
12.s1、将金源、bivo4/biocl p-n型异质结光催化剂分散于第一溶剂中,进行光还原反应,制得金纳米颗粒负载的bivo4/biocl p-n型异质结光催化剂;其中,bivo4/biocl p-n型异质结光催化剂包括biocl纳米片和负载于biocl纳米片表面的bivo4纳米颗粒;
13.s2、将所述金纳米颗粒负载的bivo4/biocl p-n型异质结光催化剂分散于第二溶剂中,加入含硫化合物搅拌混合,所述含硫化合物吸附在所述金属纳米颗粒的表面;而后加入可溶性镉盐反应,制得z型光催化剂。
14.以上制备方法中,先将金源和bivo4/biocl p-n型异质结光催化剂分散于第一溶剂中,再通过光还原法可将金源中的金离子还原为金纳米颗粒,并选择性沉积负载在bivo4/biocl p-n型异质结光催化剂中的bivo4纳米颗粒上,制得金纳米颗粒负载的bivo4/biocl p-n型异质结光催化剂;而后将其分散于第二溶剂中,加入含硫化合物,基于含硫化合物与金之间的强烈相互作用,含硫化合物吸附在金纳米颗粒的表面;而加入可溶性镉盐,其中cd
2+
可与含硫化合物水解释放的s
2-生成cds,负载在金纳米颗粒表面,从而实现在bivo4/biocl p-n型异质结光催化剂中的bivo4端通过金金属层(或金纳米颗粒)定向负载cds纳米颗粒,构建具有载流子双分离通道的z型光催化剂。
15.在本发明的一些实施方式中,步骤s1具体可包括:将bivo4/biocl p-n型异质结光催化剂分散于第一溶剂中,而后加入金源,排除氧气,并在暗态条件下搅拌混合;而后采用
可见光照射,使金源中的金离子还原为金纳米颗粒沉积负载在bivo4基异质结光催化剂中的bivo4上,而后固液分离取沉淀物,洗涤、干燥,制得金纳米颗粒负载的bivo4基异质结催化剂。其中,第一溶剂优选采用空穴清除剂溶液,根据光催化反应原理,消耗空穴(h
+
)有利于促进还原反应,因此,空穴清除剂的存在有助于促进金离子的光还原反应。空穴清除剂溶液具体可采用甲醇溶液、乙醇溶液、edta溶液等。在加入金属源之后,排除氧气,可避免催化剂在产生光子电子后,与系统中所存在氧气结合生成超氧自由基;排除氧气的方式可通入惰性气体吹扫。
16.在本发明的一些实施方式中,在步骤s1之前,还包括s0、采用bivo4纳米颗粒和盐酸溶液混合反应,制得bivo4/biocl p-n型异质结光催化剂。通过采用bivo4纳米颗粒和盐酸溶液混合反应可在bivo4纳米颗粒上原位生长biocl纳米片,以构建具有零维-二维分级纳米结构bivo4/biocl p-n异质结光催化剂。制备过程中,可通过对hcl加入量的控制,调变复合光催化剂中biocl的含量。优选地,所述bivo4/biocl p-n异质结光催化剂中biocl纳米片的含量为2.5~50%,优选为25%。通过将bivo4/biocl p-n异质结光催化剂中biocl纳米片的含量控制在以上范围,可使bivo4/biocl p-n异质结光催化剂具有较大的比表面积(大约为5~6.1m2/g)和总孔容(大约为2.7
×
10-3
~3.3
×
10-3
cm3/g),一方面基于大比表面积以提高催化剂对有机物的吸附能力,另一方面基于大总孔容,使得催化剂可以暴露更多的活性位点,从而提高光催化剂的催化性能。
17.在本发明的一些实施方式中,步骤s0之前,还包括采用反相微乳液法合成bivo4纳米颗粒;优选地,采用反相微乳液法合成bivo4纳米颗粒包括:
18.1)将铋源和钒源分别溶于酸性溶液制得水相溶液a1和水相溶液a2;
19.2)将表面活性剂、助表面活性剂、油相溶剂混合制得油相溶液b;
20.3)将所述水相溶液a1和所述水相溶液a2分别逐滴加入等量的所述油相溶液b中,混合得到混合液c1和混合液c2;
21.4)将所述混合液c1和所述混合液c2混合搅拌反应。
22.通过以上反相微乳液合成法所制备bivo4纳米颗粒粒径小,比表面积和总孔隙度大,可将bivo4纳米颗粒粒径在限定在纳米尺度,可提高bivo4的氧化活性。
23.步骤1)中,铋源可选自铋酸钠、硝酸铋、钨酸铋、硫酸铋、碱式碳酸铋、碱式水杨酸铋中的至少一种,优选采用硝酸铋;钒源可采用偏钒酸铵、五氧化二钒、三氧化二钒、偏钒酸铵、偏钒酸钠、偏钒酸钾、正钒酸钠、焦钒酸钠、硫酸氧钒、草酸氧钒、四氯化钒中的至少一种,优选采用偏钒酸铵;酸性溶液可采用硝酸溶液、柠檬酸中的至少一种。水相溶液a1和水相溶液a2的浓度可控制在0.1~0.3mol/l,优选0.2mol/l。
24.步骤2)中,表面活性剂可采用辛基苯基聚氧乙烯醚(triton x-100)、十二烷基苯磺酸钠、十二烷基磺酸钠中的一种;助表面活性剂可采用正戊醇、正己醇、正丁醇、正辛醇中的至少一种;油相溶剂可采用烷烃,如环己烷、正己烷、异辛烷中的至少一种,具体可根据微乳液配方进行选择。油相溶液b的原料可包括17.7%~42.9%表面活性剂、11.5%~28.6%助表面活性剂和28.6%~70.8%油相溶剂,优选地表面活性剂、助表面活性剂和油相溶剂的质量比为3:2:5。
25.步骤3)具体包括取等量的两份油相溶液b,而后分别将所述水相溶液a1和所述水相溶液a2分别逐滴加入两份油相溶液b中。其中,水相溶液和表面活性剂的摩尔比ω为20~
50。在该条件下,可保证制备粒径小、大小均匀、分散性好的bivo4纳米颗粒,以提升产品z型光催化剂的催化活性;且优选摩尔比ω为20。若ω过低或过高,则所获得bivo4纳米颗粒的分散程度较差,团聚明显,比表面积下降,进而会导致催化活性降低。
26.本发明的第三方面,提出了本发明第二方面所提出任一种z型光催化剂在光催化降解有机污染物和/或光解水制氢中的应用。
27.本发明的第四方面,提出了一种光催化降解有机物同步产氢的方法,包括:在可见光照射下将含有机物的废水与本发明第一方面所提出的任一种z型光催化剂混合反应;优选地,所述z型光催化剂的用量为0.1~1g/l;优选地,所述含有机物的废水中所述有机物的浓度为0.2~5mol/l;优选地,所述有机物选自有机醇类、有机醛类、有机酸类、有机胺类中的至少一种。
附图说明
28.下面结合附图和实施例对本发明做进一步的说明,其中:
29.图1为实施例1中z型光催化剂合成流程示意图;
30.图2为实施例1和对比例1、2、3、4、6、8所制得光催化剂的微观形貌测试结果图;
31.图3为实施例1和对比例4、6所制得光催化剂的xrd谱图;
32.图4为实施例1和对比例1、4、6所制得光催化剂的uv-vis drs谱图;
33.图5为实施例1和对比例4、6所制得光催化剂的xps全谱图;
34.图6为实施例1和对比例4、6所制得光催化剂的精细谱图;
35.图7为实施例1和对比例1、4、6所制得光催化剂pl光谱图;
36.图8为cds的禁带宽度及价带电位;
37.图9为实施例1所制得z型光催化剂的光催化机理示意图;
38.图10为光催化降解有机物同步产氢实验装置结构示意图;
39.图11为实施例1~8和对比例1~2所制得光催化剂的产氢活性测试结果;
40.图12为实施例1、对比例1、4、5、6、7、8所制得光催化剂的催化活性测试结果;
41.图13为有机物种类对催化降解有机物同步产氢活性的影响测试结果;
42.图14为光催化剂用量对催化降解有机物同步产氢活性的影响测试结果;
43.图15为有机物浓度对催化降解有机物同步产氢活性的影响测试结果;
44.图16为反应温度对催化降解有机物同步产氢活性的影响测试结果;
45.图17为实施例1所制得z型光催化剂的稳定性测试结构;
46.图18为实施例1所制得z型光催化剂的光催化降解乳酸同步产氢机理示意图。
具体实施方式
47.以下将结合实施例对本发明的构思及产生的技术效果进行清楚、完整地描述,以充分地理解本发明的目的、特征和效果。显然,所描述的实施例只是本发明的一部分实施例,而不是全部实施例,基于本发明的实施例,本领域的技术人员在不付出创造性劳动的前提下所获得的其他实施例,均属于本发明保护的范围。
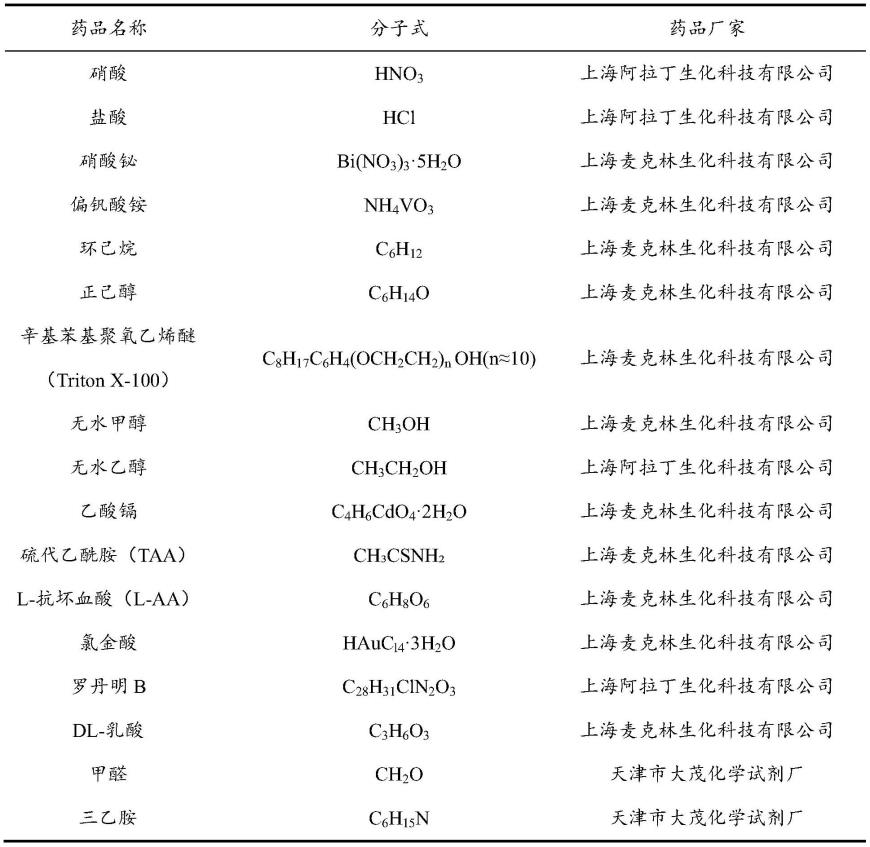
48.以下各实施例中所用到的化学药品详情如下表1:
49.表1
[0050][0051]
实施例1
[0052]
本实施例制备了一种z型催化剂,具体制备方法包括以下步骤:
[0053]
s1、采用反相微乳液法制备bivo4,包括:将bi(no3)3·
5h2o和nh4vo3分别溶于4m hno3溶液中配置为0.2m溶液,并标记为水相溶液a1和水相溶液a2;将环己烷、正己醇和triton x-100按质量比为5:2:3混合配制油相溶液b,搅拌至澄清透明溶液,持续搅拌并按照水相溶液与油相溶液中表面活性剂的摩尔比ω为20将水相溶液a1和水相溶液a2分别逐滴加入等量的两份油相溶液b中,搅拌10min获得澄清透明的无色混合液c1和黄色混合液c2,将混合液c1缓慢加入至混合液c2中,并持续搅拌反应30min,以保证反应充分,得到含有bivo4的微乳液。
[0054]
s2、bivo4/biocl p-n异质结光催化剂制备,包括:将步骤s1所制得含有bivo4的微乳液加入一定量的hcl溶液,使cl-和bi
3+
的摩尔比为1:0.25,搅拌30min使反应充分,室温下静置陈化24h,制得具有零维(0d)-二维(2d)分级纳米结构的bivo4/biocl p-n异质结光催化剂。
[0055]
s3、利用光还原法制备au负载bivo4/biocl光催化剂(即au-bivo4/biocl光催化剂),包括:将100mg步骤s2所制得的bivo4/biocl p-n异质结光催化剂超声分散在100ml甲醇水溶液(甲醇与水的体积比为1:4)中,按au在最终产品z型光催化剂中的理论含量为
2wt%,加入0.4ml的10g/l的haucl4水溶液,通入n2吹扫,并在黑暗条件下保持搅拌30min,随后使用300w氙灯光源(携420nm紫外截止滤光片)照射30min,利用光生电子将au
3+
还原为au单质纳米颗粒沉积在在bivo4端,反应结束后离心收集催化剂,使用无水乙醇和去离子水清洗数次,于60℃烘箱中干燥8h获得au-bivo4/biocl光催化剂。
[0056]
s4、制备cds-au-bivo4/biocl z型光催化剂,包括:将100mg步骤s3所制得的au-bivo4/biocl光催化剂分散在8ml无水乙醇中,加入1ml ch3csnh2乙醇溶液(1m),在黑暗条件下搅拌30min以实现ch3csnh2在au纳米颗粒上的选择性吸附,加入1mlcd(ch3coo)2·
2h2o乙醇溶液(1m)搅拌30min,得到混合液,其中镉源和硫源的摩尔浓度为0.1mol/l;再离心收集催化剂,并置于阴凉通风处干燥后获得cds-au-bivo4/biocl z型光催化剂,其中cds的理论含量为10%。
[0057]
以上步骤s3~s4中,通过在具有0d-2d分级纳米结构bivo4/biocl p-n异质结光催化剂的bivo4端定向负载cds合成z型光催化剂,其具体合成方案如图1所示。首先,在步骤1中,以haucl4为au源,甲醇(ch3oh)作为h
+
清除剂,利用bivo4的导带上的e-将au
3+
还原为au单质选择性沉积在bivo4纳米颗粒上,而位于biocl价带上的h
+
将甲醇氧化为co2和h2o,以上过程所发生反应包括下式(1)、(2)、(3),反应制得au负载的bivo4/biocl光催化剂(au-bivo4/biocl光催化剂),之后在制备cds-au-bivo4/biocl z型光催化剂过程中,首先得益于含硫化合物与金表面之间的强烈相互作用,ch3csnh2优先吸附在au纳米粒子的表面;cds生成于步骤3中,当ch3csnh2逐渐水解并释放s
2-,s
2-与cd
2+
反应生成cds,如下式(4)所示。
[0058]
bivo4/biocl+hυ
→
e-‑
bivo4+h
+-biocl
ꢀꢀꢀ
(1)
[0059]
au
3+
+e-‑
bivo4→
au-bivo4ꢀꢀꢀ
(2)
[0060]
ch3oh+h
+-biocl
→
co2+h2o
ꢀꢀꢀ
(3)
[0061]
cd
2+
+s
2-‑
au-bivo4→
cds-au-bivo4ꢀꢀꢀ
(4)
[0062]
实施例2
[0063]
本实施例制备了一种z型催化剂,本实施例与实施例1的区别在于:步骤s3中,按au在最终产品z型光催化剂中的理论含量为1wt%,将haucl4水溶液的用量调整为0.2ml,其他操作与实施例1相同,制得cds-au-bivo4/biocl z型催化剂。
[0064]
实施例3
[0065]
本实施例制备了一种z型催化剂,本实施例与实施例1的区别在于:步骤s3中,按au在最终产品z型光催化剂中的理论含量为1.5wt%,将haucl4水溶液的用量调整为0.3ml,其他操作与实施例1相同,制得cds-au-bivo4/biocl z型催化剂。
[0066]
实施例4
[0067]
本实施例制备了一种z型催化剂,本实施例与实施例1的区别在于:步骤s3中,按au在最终产品z型光催化剂中的理论含量为2.5wt%,将haucl4水溶液的用量调整为0.5ml,其他操作与实施例1相同,制得cds-au-bivo4/biocl z型催化剂。
[0068]
对比例1
[0069]
本对比例1制备了一种光催化剂,本对比例与实施例1的区别在于:本对比例取消步骤s3的操作;步骤s4中,采用等量步骤s2所制得的bivo4/biocl p-n异质结光催化剂代替au-bivo4/biocl光催化剂,其他操作与实施例1相同。本对比例所制得产品催化剂为cds-bivo4/biocl光催化剂。
[0070]
实施例5
[0071]
本实施例制备了一种z型催化剂,本实施例与实施例1的区别在于:步骤s4中,将分散溶剂无水乙醇的用量由实施例1中的10ml调整为9ml;将所采用ch3csnh2乙醇溶液(1m)和cd(ch3coo)2·
2h2o乙醇溶液(1m)的用量由实施例1中的1ml均调整为0.5ml,所得混合液中镉源和硫源的摩尔浓度为0.05mol/l;其他操作与实施例1相同,制得cds-au-bivo4/biocl z型催化剂,其中,cds的理论含量为5%。
[0072]
实施例6
[0073]
本实施例制备了一种z型催化剂,本实施例与实施例1的区别在于:步骤s4中,将分散溶剂无水乙醇的用量由实施例1中的10ml调整为7ml;将所采用ch3csnh2乙醇溶液(1m)和cd(ch3coo)2·
2h2o乙醇溶液(1m)的用量由实施例1中的1ml均调整为1.5ml,所得混合液中镉源和硫源的摩尔浓度为0.15mol/l;其他操作与实施例1相同,制得cds-au-bivo4/biocl z型催化剂,其中,cds的理论含量为15%。
[0074]
实施例7
[0075]
本实施例制备了一种z型催化剂,本实施例与实施例1的区别在于:步骤s4中,将分散溶剂无水乙醇的用量由实施例1中的10ml调整为6ml;将所采用ch3csnh2乙醇溶液(1m)和cd(ch3coo)2·
2h2o乙醇溶液(1m)的用量由实施例1中的1ml均调整为2ml,所得混合液中镉源和硫源的摩尔浓度为0.2mol/l;其他操作与实施例1相同,制得cds-au-bivo4/biocl z型催化剂,其中,cds的理论含量为20%。
[0076]
对比例2
[0077]
本对比例制备了一种光催化剂,本对比例与实施例1的区别在于:本对比例取消了实施例1中步骤s4的操作,其他操作与实施例1相同,以实施例1中步骤s3所制得au-bivo4/biocl光催化剂作为产品光催化剂。
[0078]
对比例3
[0079]
本对比例制备了一种光催化剂,其制备方法包括:
[0080]
s1、按与实施例1中步骤s1相同的操作制得含bivo4的微乳液;而后将含有bivo4的微乳液在室温下静置陈化48h后,加入过量的体积比为1:1的乙醇水溶液直至沉淀完全析出,离心将沉淀分离,分别用无水乙醇、去离子水冲洗数次,于60℃烘箱中干燥8h获得bivo4前驱体;将获得的bivo4前驱体置于马弗炉中,以5℃/min为升温速率升温至500℃并保持1h,获得bivo4纳米颗粒,即光催化剂。
[0081]
对比例4
[0082]
本对比例制备了一种光催化剂,本对比例与实施例1的区别在于:本对比例取消了实施例1中步骤s3、s4的操作,其他操作与实施例1相同,以实施例1中步骤s2所制得bivo4/biocl光催化剂作为产品光催化剂。
[0083]
对比例5
[0084]
本对比例制备了一种光催化剂,其制备方法包括:
[0085]
s1、按与实施例1中步骤s1相同的操作制得含bivo4的微乳液;而后将含有bivo4的微乳液在室温下静置陈化48h后,加入过量的体积比为1:1的乙醇水溶液直至沉淀完全析出,离心将沉淀分离,分别用无水乙醇、去离子水冲洗数次,于60℃烘箱中干燥8h获得bivo4前驱体;将获得的bivo4前驱体置于马弗炉中,以5℃/min为升温速率升温至500℃并保持
1h,最终获得bivo4纳米颗粒。
[0086]
s2、取100mg步骤s1所制得的bivo4纳米颗粒代替实施例1中步骤s4中的au-bivo4/biocl光催化剂,而后按与实施例1中步骤s4相同的操作,制得cds-bivo4光催化剂。
[0087]
对比例6
[0088]
本对比例制备了一种光催化剂,其制备方法包括:采用1ml ch3csnh2乙醇溶液(1m)与1ml cd(ch3coo)2·
2h2o乙醇溶液(1m)混合搅拌30min后离心收集催化剂,并置于阴凉通风处干燥后获得cds光催化剂。
[0089]
对比例7
[0090]
本对比例制备了一种光催化剂,其制备方法包括:将100mg biocl纳米片分散在10ml无水乙醇中,加入1ml ch3csnh2乙醇溶液(1m)、1ml cd(ch3coo)2·
2h2o乙醇溶液(1m)搅拌30min后离心收集催化剂,并置于阴凉通风处干燥后获得cds-biocl光催化剂。
[0091]
对比例8
[0092]
本对比例制备了一种光催化剂,本对比例与实施例1的区别在于:本对比例的步骤s3中利用化学还原法制备au负载bivo4/biocl光催化剂(即au(c)-bivo4/biocl光催化剂),具体包括:100mg bivo4/biocl p-n异质结光催化剂超声分散在100ml去离子水中,并加入0.1mmol l-抗坏血酸,搅拌10min后向溶液中加入与0.4ml的10g/l的haucl4水溶液,搅拌30min后离心收集催化剂,经无水乙醇和去离子水清洗数次,于60℃烘箱中干燥8h获得au(c)-bivo4/biocl光催化剂。其他操作与实施例1相同,本对比例所制得产品催化剂为cds-au(c)-bivo4/biocl光催化剂。
[0093]
性能测试
[0094]
(一)催化剂表征
[0095]
1、形貌及微观结构分析
[0096]
采用扫描电子显微镜(生产厂家:日本日立公司,型号:su1080)和透射电子显微镜(生产厂家:日本日立公司,型号:tecnai g2 f30)分别对实施例1、对比例1、2、3、4、6、8所制得催化剂的微观形貌进行测试分析,所得微观形貌测试结果包括图2所示。图2中(a)表示对比例6所制得cds光催化剂的sem图,尺度为500nm;(b)表示对比例3所制得bivo4光催化剂的sem图,尺度为500nm;(c)、(d)表示对比例4所制得bivo4/biocl光催化剂的sem图,尺度分分别为2.0μm、1.0μm;(e)表示对比例1所制得cds-bivo4/biocl的sem图,尺度为500nm;(f)表示对比例8所制得cds-au(c)-bivo4/biocl光催化剂的sem图,尺度为500nm;(g)、(h)分别表示对比例2所制得au-bivo4/biocl光催化剂的sem图和tem图;尺度分别为500nm、100nm;(i)、(j)分别表示实施例1所制得cds-au-bivo4/biocl光催化剂的sem图和tem图,尺度分别为500nm、100nm。
[0097]
由图2中(a)所示cds光催化剂的sem图可以看出,cds纳米颗粒粒径约为20nm。由图2中(b)所示bivo4光催化剂的sem图可以看出,对比例3所制得bivo4纳米颗粒间分散性良好,颗粒粒径均匀分布,分布于80~100nm之间。由图2中(c)、(d)所示bivo4/biocl光催化剂的sem图可以看出,催化剂中含有明显的纳米片分布,在纳米片表面均匀分布纳米颗粒,且纳米颗粒与纳米片之间紧密结合,分散均匀。由图2中(e)所示cds-bivo4/biocl的sem图可以看出,cds纳米颗粒均匀分布在bivo4和biocl;由图2中(f)所示cds-au(c)-bivo4/biocl光催化剂的sem图也可以观察到,cds纳米颗粒均匀分布在bivo4和biocl,由此证明,对比例8中
使用化学还原法沉积au无法实现cds的选择性负载。从图2中(g)所示au-bivo4/biocl光催化剂的sem图,可以观察到biocl纳米片表面光滑无物质沉积,而在bivo4纳米颗粒上可以观察到有物质沉积,而图2中(h)所示分别率更高的tem图进一步证明了这一现象。由图2中(i)所示cds-au-bivo4/biocl光催化剂的sem图可以看出,实施例1中在负载cds后,biocl纳米片表面依旧光滑,而bivo4纳米颗粒表面可以清楚观察的有cds的存在,通过图2中(j)所示tem图可发现bivo4纳米颗粒表面为分散的cds纳米颗粒。另外,为了进一步证明au和cds的存在,使用透射电镜配备的eds设备分析图2中(j)所圈出区域内的元素成分,所得结果如图2中(k)所示,所得结果显示此处存在bi、o、v、cd、s和au而没有cl的存在。通过以上分析,可以认为实施例1中成功地将cds选择性负载在了bivo4/biocl光催化剂中的bivo4端。
[0098]
2、晶相结构分析
[0099]
采用d/max 2550vb/pc型x射线衍射仪(xrd)分别对实施例1所制得cds-au-bivo4/biocl光催化剂、对比例4所制得bivo4/biocl光催化剂和对比例6所制得cds光催化剂的晶相结构进行测定,测试条件为cu阳极靶,kα射线,加速电压45kv电流50ma,扫描角度为5-90
°
,扫面速率5
°
/min,所得结果如图3所示。
[0100]
从图3可以看出,cds的xrd谱峰与立方相cds(jcpds文件:89-0440)匹配良好,谱图中26.5
°
、43.9
°
以及52.0
°
处观察到特征衍射峰,分别对应立方相cds的(111)、(220)和(311)晶面。在cds-au-bivo4/biocl光催化剂的谱图中,可明显观察到bivo4和biocl的特征峰,此外,在26.5
°
处也可明显观察到cds(111)晶面的谱峰,表明bivo4/biocl与cds成功复合。由于cds-au-bivo4/biocl光催化剂中au的含量较少,并未在其xrd谱图中观察到au的特征衍射峰。
[0101]
3、吸收性能分析
[0102]
采用uv-2450型紫外-可见漫反射仪(uv-vis drs)对实施例1所制得cds-au-bivo4/biocl光催化剂、对比例1所制得cds-bivo4/biocl光催化剂、对比例4所制得bivo4/biocl光催化剂和对比例6所制得cds光催化剂的吸光性能进行测试,测试过程中以baso4为参比,测试波长范围为200-800nm,所得结果如图4所示。
[0103]
由图4可知,相较于bivo4/biocl光催化剂,纯cds光催化剂展示出了更高的吸光强度,但其吸光范围较窄,吸收边位于501nm,cds-bivo4/biocl光催化剂的吸光范围和吸收强度具有显著提升,表明bivo4/biocl和cds复合可以有效提升对光的利用效率。在cds-au-bivo4/biocl光催化剂谱图中的620nm处,可明显观察到一处肩峰,可归应于au纳米颗粒的等离子体共振(spr)吸收,表明光催化剂中au的存在。同时,该催化剂相比于cds-bivo4/biocl光催化剂表现出更宽的吸收边界和更高的吸光强度,这可能是由于紧密接触的au和cds的共同作用。由此可见,实施例1中au的加入不仅作为锚定点使cds定向负载在bivo4端,还使催化剂的吸光性能得以进一步提升,这对提升催化剂活性显著有利。
[0104]
4、元素组成及化学价态分析
[0105]
采用在配备有在250w操作的al kα辐射源的thermo scientific k-alpha型x射线光电子能谱仪(xps)分别对实施例1所制得cds-au-bivo4/biocl光催化剂、对比例4所制得bivo4/biocl光催化剂和对比例6所制得cds光催化剂的元素组成及化学价态,所得结果如图5和图6所示,图6所示xps精细谱中,(a)为bi 4f,s 2p谱图;(b)为cd 3d谱图;(c)为v 2p谱图;(d)为cl 2p谱图;(e)为au 4f谱图;(f)为o1s谱图。
[0106]
图5中cds-au-bivo4/biocl的全谱图证实了bi、v、o、cl和cd、s以及au元素共存。由于s 2p谱峰和bi 4f谱峰存在重合现象,图6中(a)同时展示了s 2p和bi 4f的高分辨xps谱图,对于bivo4/biocl和cds-au-bivo4/biocl光催化剂,159.0ev或159.3ev和164.3ev或164.7ev结合能处的谱峰分别归属于bi 4f
7/2
和bi 4f
5/2
,表明bi的存在形式为bi
3+
;cds-au-bivo4/biocl和cds光催化剂谱图中,162.3ev或162.5ev和164.7ev或164.8ev结合能处的谱峰对应s 2p
3/2
和s 2p
1/2
,表明s的存在形式为s
2-。图6中(b)展示了cds-au-bivo4/biocl和cds光催化剂的cd 3d的高分辨xps谱图,405.2ev或405.4ev和411.9ev或412.1ev处分别出现了cd 3d
5/2
和cd 3d
3/2
的峰,表明cd在催化剂中的存在形式为cd
2+
。bivo4/biocl和cds-au-bivo4/biocl的v 2p和cl 2p谱图分别展示在图6中(c)和(d),v 2p
3/2
和v 2p
1/2
谱峰分别位于516.7ev或517.0ev和524.5ev或524.8ev处,cl 2p
3/2
和cl 2p
1/2
谱峰分别位于197.9ev或198.4ev和199.2ev或199.9ev处。
[0107]
值得注意的是,对比bivo4/biocl和cds光催化剂的谱图,cds-au-bivo4/biocl中的bi 4f、v 2p和cl 2p元素的谱峰均向高结合能方向发生偏移,cd 3d和s 2p元素的谱峰则是向低结合能方向发生偏移。这一现象表明复合光催化剂材料中的不同组分之间存在强烈的电子相互作用。结合能的正移归因于电子密度的降低,而结合能的负移则表明电子密度的增加,证明复合体系中电子向cds转移。同时,图6中(e)展示了cds-au-bivo4/biocl的au 4f谱图,84.2ev和88.3ev处的谱峰归属于单质金的au 4f
7/2
和au 4f
5/2
。图6中(f)所示,bivo4/biocl和cds-au-bivo4/biocl的o1s谱峰均可分裂为529.9ev或530.6ev处的晶格氧(o
l
)和531.5ev或532.0ev处的化学吸附氧(oc)。bivo4/biocl中oc/o
l
为0.53,而cds-au-bivo4/biocl的oc/o
l
提升至1.07,证明复合光催化剂体系的形成极大促进了氧空位的形成。
[0108]
5、光生载流子迁移和复合速率测定
[0109]
采用horiba fm4型光致发光光谱仪(pl)测定实施例1所制得cds-au-bivo4/biocl光催化剂、对比例1所制得cds-bivo4/biocl光催化剂、对比例4所制得bivo4/biocl光催化剂和对比例6所制得cds光催化剂的光生载流子的复合分离效率,所得结果如图7所示。
[0110]
图7中各光催化剂的光致发光强度由大到小依次为cds、bivo4/biocl、cds-bivo4/biocl和cds-au-bivo4/biocl,cds-au-bivo4/biocl最低的光致发光强度,表明所获得的光催化剂具有优异的光生载流子分离性能,可以保证催化剂具有良好的催化活性。此外,根据cds-bivo4/biocl光催化剂的pl光谱,可以明显看出在bivo4/biocl光催化剂上负载cds有效抑制了光生载流子的复合,但其分离效率远不及cds-au-bivo4/biocl,说明依据光生载流子的迁移路径进行异质结的构建可进一步提升光生载流子分离效率。
[0111]
(三)z型光催化剂的光催化机理
[0112]
cds的禁带宽度及价带电位如图8所示,其中(a)为cds的禁带宽度,(b)为cds的价带电位,可以得到cds的禁带宽度为2.61ev,价带电位处于1.30ev,由此可得到其导带电位为-1.31ev,其能带结构图如图9中所示。并结合上文中的xps分析(即元素组成及化学价态分析)可知,在cds-au-bivo4/biocl光催化剂中,cds呈现得电子状态,bivo4/biocl则表现为失电子状态,表明电子由bivo4/biocl向cds发生转移。由此可以判断,cds-au-bivo4/biocl z型光催化剂的光催化机理如图9所示,当bivo4和cds受到可见光激发后,各自产生光生电子-空穴对,e-跃迁至导带(cb),h
+
留在价带(vb)上。bivo
4 cb上的e-经过bivo4和cds之间的电子中介体au转移到cds的vb上与其产生的h
+
发生复合,au的存在降低了e-的传输阻抗,提
高界面上的e-传输速率,因此,cds cb上的e-被保留用于h2的产生,而bivo
4 vb上的h
+
则进一步转移并被保留在biocl的vb上,形成
·
oh或直接参与氧化反应,实现了光生载流子的双通道分离。
[0113]
(四)光催化剂催化活性评价
[0114]
使用以上所制得各光催化剂进行光催化降解有机物同步产氢实验,依据催化剂的h2产率和toc去除率(或反应后与反应前溶液中的toc含量之比)对光催化剂进行催化活性评价。
[0115]
产氢及降解所用实验装置为湖南华思仪器公司生产的标准全自动光解水制氢循环反应系统,如图10所示,包括光源10、反应系统、循环冷却装置30以及气路系统组成;光源10采用北京泊菲莱科技有限公司pls-sxe 300+型号氙灯光源系统,携420nm紫外截止滤光片11用于滤除紫外光,其位于反应液面上方约15cm处。反应系统由反应器21与恒温磁力搅拌器22组成,反应器21由不锈钢材质构成,容积为300ml,反应器21上布设有气体采样口和液体采样口211。循环冷却系统30通过将冷却水循环流过反应器21外壁及其两端气路冷凝管,保证反应在恒定温度下进行,并保证水蒸气不进入气相色谱进样口。气路系统由气瓶41及管路42组成,气体由管路42进入反应器21,待反应结束后,反应器内气体进入气相色谱仪50进行组分测试。
[0116]
具体测试方法包括:将50mg光催化剂分散在100ml一定浓度有机污染物的(乳酸、甲醇、甲醛和三乙胺)反应液中,保持暗反应30min,在此过程中,用n2吹扫反应器,以除去反应器中的氧气;达到吸附-解吸平衡后,使用300w氙灯(携紫外截止滤光片)照射悬浮物;在反应过程中,每隔15min取1ml气相样品,通过密闭管路送至气相色谱仪检测h2含量;与此同时,从液体采样口采集2ml液相产物,利用总有机碳检测器测定溶液中的toc含量。然后通过以下公式(ⅰ)计算出产氢效率,通过公式(ⅱ)计算出toc去除效率,和/或通过公式(ⅲ)计算出反应后与反应前溶液中的toc含量之比,以评价光催化剂活性。
[0117][0118]
式中,表示产氢速率,n表示氢气的物质的量(μmol),t表示反应时间(h),m表示光催化剂的质量(g);
[0119][0120]
式中,t表示toc去除率,t0表示反应前溶液中toc含量(mg/l),t
t
表示反应后溶液中toc含量(mg/l);
[0121][0122]
式中,m表示反应后与反应前溶液中的toc含量之比,t0表示反应前溶液中toc含量(mg/l),t
t
表示反应后溶液中toc含量(mg/l)。
[0123]
具体催化活性评价测试包括:
[0124]
1、不同au含量和cds含量对光催化剂的产氢活性的影响
[0125]
采用以上方法,对实施例1~7和对比例1~2所制得光催化剂的催化活性进行评价测试,其中以体积分数为10%的乳酸水溶液(即dl-乳酸水溶液)作为反应液,具体通过测定
产氢效率进行催化活性评测,所得结果如图11所示,图11中(a)为实施例1~4和对比例1不同au的含量下光催化剂的产氢活性测试结果,(b)为实施例1、5~7和对比例2不同cds含量下光催化剂的产氢活性测试结果。
[0126]
由图11中(a)可知,au的含量影响光催化剂的产氢活性,具体地,光催化剂的产氢活性随au的含量先增大后减小,当au的负载量为2wt%时,cds-au-bivo4/biocl具有最佳产氢活性,h2产率为1936μmol
·
g-1
·
h-1
。当催化剂中au的含量过小时,cds无法被完全锚定在bivo4端,当au含量过大时,au纳米颗粒粒径增大,可能会遮挡bivo4/biocl的活性位点,导致活性下降。通过icp-oes准确定量实施例1所制得z型催化剂中au的含量,结果显示为1.85wt%。
[0127]
由图11中(b)可知,光催化剂的产氢活性还与其中的cds的含量密切相关,随着cds含量的增加,光催化剂产氢活性呈现先增大后减小的趋势。当cd源与s源浓度为0.1mol/l时,获得的光催化剂具有最大产氢速率为2257μmol
·
g-1
·
h-1
。当cds含量持续增大,产氢活性反而降低,可能是因为过多的cds纳米颗粒覆盖在bivo4/biocl上,降低了bivo4/biocl对光的利用率,同时阻碍了其与底物的反应。使用icp-oes对实施例1中cd源与s源浓度均控制在0.1mol/l(即0.1m)下所制得z型光催化剂中的cd元素含量进行测定以确定催化剂中cds的含量,结果显示为10.95wt%。
[0128]
2、不同类型光催化剂的催化活性
[0129]
采用以上方法,对实施例1的cds-au-bivo4/biocl、对比例8的cds-au(c)-bivo4/biocl、对比例1的cds-bivo4/biocl、对比例4的bivo4/biocl、对比例5的cds-bivo4、对比例7的cds-biocl和对比例6cds光催化剂的催化活性进行评价测试,具体地,以体积分数为10%的乳酸水溶液作为反应液,通过分别测定产氢效率(即不同时间下的产氢量)、以及反应后与反应前溶液中的toc含量之比以对各光催化剂的催化产氢活性和催化降解有机物活性进行评测,所得结果如图12所示,图12中(a)为催化产氢活性测试结果;(b)为催化降解有机物活性测试结果。
[0130]
经测试得出,单一cds因其容易发生光腐蚀,导致其活性较差,其h2产率仅为623μmol
·
g-1
·
h-1
,toc去除率为6.17%(2h)。当cds与单一bivo4复合后,催化剂活性具有一定提升,cds-bivo4的h2产率为791μmol
·
g-1
·
h-1
,toc去除率为12.58%。cds-biocl虽然显示出比cds更高的矿化率(9.45%),但其产氢活性却低于cds,这可能是因为biocl无法被可见光激发产生光生电子,因此不能通过载流子的分离抑制cds的光腐蚀。cds-bivo4/biocl相比于cds-bivo4和cds-biocl活性具有显著提升,其h2产率为1460μmol
·
g-1
·
h-1
,toc去除率为16.47%(2h),可见,当cds被引入复合体系后,bivo4和biocl依旧存在相互作用,且有助于提升整体催化剂的产氢和降解活性。
[0131]
通过对比分析cds-au-bivo4/biocl、cds-au(c)-bivo4/biocl和cds-bivo4/biocl光催化剂的活性评价结果,可以看出au的加入可以提升光催化剂的活性,但提升程度与au的负载方式显著相关。结合前文对催化剂吸光性能和载流子复合效率的分析可知,当au以光还原沉积的方式负载在bivo4/biocl的bivo4端时,不仅可以显著提升光催化剂的吸光性能,更有利于提升光生载流子的迁移速率,这对于提升光催化剂活性是至关重要的。cds-au-bivo4/biocl具有最佳活性,h2产率为2420μmol
·
g-1
·
h-1
,是单一cds的3.88倍,toc去除率为21.67%(2h),是bivo4/biocl的1.44倍。
[0132]
3、有机物种类对催化降解污染物同步产氢活性的影响
[0133]
采用实施例1所制得cds-au-bivo4/biocl光催化剂,选择体积分数为10%的甲醇(ch3oh)溶液、甲醛(hcho)溶液、乳酸(ch3ch(oh)cooh)溶液和三乙胺((ch3ch2)3n)溶液分别作为有机醇类、有机醛类、有机酸类以及有机胺类的代表物质,并作为反应液,按照类似于以上的方法分别进行光催化降解有机物同步产氢研究试验,以考察不同种类有机物添加下光催化剂的催化剂活性,具体通过分别测定产氢效率(即不同时间下的产氢量)和反应2h后的toc去除率以对光催化剂的催化产氢活性和催化降解有机物活性进行评测,所得结果如图13所示,图13中(a)为催化产氢活性测试结果;(b)为催化降解有机物活性测试结果。
[0134]
由图13中(a)可知,以上不同有机物添加下光催化剂的产氢活性由大到小分别为乳酸、三乙胺、甲醛和甲醇,产氢速率分别为2420μmol
·
g-1
·
h-1
、679μmol
·
g-1
·
h-1
、309μmol
·
g-1
·
h-1
、176μmol
·
g-1
·
h-1
。图13中(b)展示了各类有机物经过2h反应后的矿化率,乳酸为21.67%,三乙胺为22.08%,甲醛为15.32%,甲醇为30.49%。以乳酸作为反应底物,其产氢活性要显著高于其他有机物,主要时因为其对空穴的消耗能力要显著强于其他有机物。不同有机物toc去除率的差异主要源于各类物质的矿化速率不同导致,这与有机物各自的降解路径密切相关。
[0135]
5、光催化剂用量对催化降解污染物同步产氢活性的影响
[0136]
按照类似以上方法,在25℃条件下,以体积分数为10%的乳酸水溶液作为反应液,通过在反应液中加入不同量(0.1g/l、0.25g/l、0.5g/l、0.75g/l和1g/l)的催化剂进行催化活性评价实验,以探究催化剂用量对h2产率和toc去除率的影响,所得结果如图14所示,图14中(a)为催化产氢活性测试结果;(b)为催化降解有机物活性(即反应2h后的toc去除率)测试结果。
[0137]
由图14中(a)可以看出,随着光催化剂用量的增加,h2产率呈现先增大后减小趋势,当光催化剂用量增加到0.5g/l时,得到最大h2产率为3016μmol
·
g-1
·
h-1
。图14中(b)显示的toc去除率与h2产率表现出相同变化趋势,也是随着光催化剂用量增大而先增大后减小,催化剂用量为0.5g/l时得到最大值23.70%。当光催化剂用量过少时,光催化剂对光的利用不充分,光催化剂用量在0.5g/l以内催化活性与光催化剂用量正相关,随着光催化剂的用量增加,可用于光催化反应的活性中心随之增多,能够提升光催化活性。但是当光催化剂用量过多时,光催化剂颗粒通过对光的吸收和散射降低了光的辐射深度,处于底部的光催化剂颗粒无法被激发而导致催化活性降低。因此,光催化剂的用量优选0.5g/l。
[0138]
6、有机物浓度对催化降解污染物同步产氢活性的影响
[0139]
按照类似以上方法,在25℃条件下,控制光催化剂用量为0.5g/l,以不同浓度的乳酸水溶液(0.2mol/l、0.5mol/l、1mol/l、2mol/l和5mol/l)作为反应液进行光催化降解有机物同步产氢实验,以探究有机物浓度对h2产率和toc去除率的影响,所得结果如图15所示,图15中(a)为催化产氢活性测试结果;(b)为催化降解有机物活性(反应2h后的toc去除率)测试结果。
[0140]
由图15中(a)所示,随着乳酸初始浓度增大,光催化剂的产氢活性随之提升。值得注意的时,当乳酸浓度小于1mol/l时,随着乳酸浓度的增大,产氢活性随之快速提升,而当乳酸浓度大于1mol/l后,产氢活性的提升则显著变慢,即使将乳酸浓度提升至5mol/l,也只有小幅度的提升。分析原因可能是在低浓度时,光催化剂表面未达到污染物吸附饱和,此时
随着污染物浓度增大,反应愈发充分,当污染物浓度超过1mol/l时,光催化剂表面活性位点已被充分利用,继续增大浓度对产氢活性提升较小。乳酸初始浓度对催化剂降解活性的影响如图15中(b)所示,光催化剂在初始浓度为0.2mol/l、0.5mol/l、1mol/l、2mol/l和5mol/l的乳酸溶液中反应2h后,toc去除率分别为45.90%、28.89%、25.09%、14.09%和10.55%。虽然,光催化剂对有机污染物的矿化率呈现下降趋势,但是光催化剂所去除toc的绝对值是增大的,分别为17.15mmol、26.34mmol、43.57mmol、50.42mmol和93.18mmol。由此可见,随着乳酸初始浓度增大,光催化剂的产氢和降解活性均随之增大,且当乳酸浓度大于1mol/l后,活性提升效果不明显,且使用过大有机物浓度会导致资源浪费,因此,乳酸初始浓度以选取1mol/l为益。
[0141]
7、反应温度对催化降解污染物同步产氢活性的影响
[0142]
控制光催化剂用量为0.5g/l,乳酸溶液的浓度为1mol/l,分别在25℃、35℃、45℃、55℃条件下按照类似以上方法进行光催化降解有机物同步产氢研究,具体测试光催化剂的产氢效率和反应2h后的toc去除率,以探究反应温度对h2产率和toc去除率的影响,所得结果如图16所示,图16中(a)为催化产氢活性测试结果;(b)为催化降解有机物活性测试结果。
[0143]
由图16所示测试结果可以发现,随着反应温度升高,氢气产率缓慢升高,当反应温度由25℃提升至55℃时,产氢速率提升392μmol
·
g-1
·
h-1
,降解活性与产氢活性变化规律一致,随着温度升高,toc去除率提升3.08%。由此可见,升高温度对光催化反应效率提升效果有限,并且维持反应在高温条件下会产生额外能耗,因此优选室温25℃作为反应温度。
[0144]
7、光催化剂的稳定性测试
[0145]
固定反应温度为25℃,光催化剂用量为0.5g/l,乳酸初始浓度为1mol/l,采用实施例1所制得光催化剂,按照类似以上方法测试光催化剂的产氢效率和反应2h后的toc去除率,以进行催化活性评测,并通过循环实验判断催化剂的稳定性。具体在每次实验结束后,将光催化剂与反应液分离,并使用超纯水和无水乙醇清洗数次,然后将洗净的光催化剂分散在重新配置的反应液中,进行重复实验。所得结果如图17所示,图17中(a)为催化产氢稳定性测试结果;(b)为催化降解有机物稳定性测试结果。
[0146]
由图17中(a)可知,经历5次循环实验后,h2产率为2921μmol
·
g-1
·
h-1
,降解活性如图17中(b)所示,最后一次实验toc去除率为21.43%。由此可见,光催化剂的活性没有发生明显下降,可以判断光催化剂性质稳定。在实际应用中,光催化剂的安全性也是一项重要指标。使用icp-ms检测循环实验后水溶液中bi和cd元素含量分别为2.63
×
10-4
mg/l和2.80mg/l,由此可见z型光催化剂具有较好的稳定性和安全性。
[0147]
(五)z型光催化剂的降解有机污染物同步产氢机理
[0148]
发明人为探明在实施例1所制得z型光催化剂光催化降解乳酸同步产氢研究过程中乳酸(ch3ch(oh)cooh)的降解路径,采用液相色谱-质谱(lc-ms/ms)鉴定乳酸降解过程中的中间产物,共发现有三种子离子,相对分子质量分别为44.05、46.07和60.05,对比可知这三种物质分别为乙醛(ch3cho)、乙醇(ch3ch2oh)和乙酸(ch3cooh)。此过程主要是ch3ch(oh)cooh脱羧降解生成乙醇,乙醇在催化剂催化反应下被迅速氧化为乙醛,同时乙醛逐步被氧化为乙酸,随着反应时间的增加,乙酸不断被氧化分解为co2和h2o,使乳酸浓度明显降低,这与toc的测定结果相符。
[0149]
结合前文中对z型光催化剂催化剂的光催化机理的分析,推断其光催化降解乳酸
同步产氢机理如图18所示。首先,在可见光照射下,bivo4和cds同时被激发产生光生载流子,e-迁移至半导体导带(cb)上,h
+
则留在半导体价带(vb)上。此时,载流子依照两条通道进行迁移,bivo
4 cb上的e-通过au与迁移至cds vb上与空穴发生复合,使得cds cb上的电子得以保留并用于还原h
+
获得h2;另一条通道为bivo
4 vb上的h
+
通过bivo4和biocl间紧密接触的界面转移至biocl的vb上,用于
·
oh的产生或直接进行乳酸的氧化降解反应。在光催化降解乳酸同时产氢反应的每一步中均存在h2o分子和有机物分子在催化剂表面的双吸附过程,ch3ch(oh)cooh、ch3ch2oh、ch3cho、ch3cooh不断吸附在催化剂表面发生氧化反应,最终实现有机物的彻底矿化。第
①
步,ch3ch(oh)cooh分子吸附催化剂上,ch3ch(oh)cooh受到h
+
和
·
oh的氧化作用,进行脱羧反应产生ch3ch2oh;第
②
步,ch3ch2oh分子吸附至催化剂表面,继续受h
+
和
·
oh作用,发生氧化反应生成ch3cho;第
③
步,ch3cho分子吸附至催化剂表面,并进一步发生氧化反应生成ch3cooh,根据以上有机物种类对催化降解污染物同步产氢的影响研究,醛类物质在相同时间内的toc去除率最低,因此ch3cho的氧化反应速率较慢,成为该反应过程中的受控步骤;第
④
步,ch3cooh分子吸附至催化剂表面并氧化分解成co2和h2o,实现对乳酸的彻底矿化;同时,h2由溶液中的h
+
受e-还原作用生成,这一反应发生在每一步中,由此,在光催化降解ch3ch(oh)cooh的同时实现了光催化产氢。
[0150]
以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对本发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1
- 一种对双酚a具有高矿化率的可见光响应型复合光催化剂的制作方法
- 多级式光催化臭氧氧化反应器及其光催化剂的制备方法
- 一种z型光催化剂及其制备方法
- 抗病毒性组合物、抗病毒剂、光催化剂以及病毒灭活方法
- 一种复合可见光光催化剂Ag<sub>2</sub>CO<sub>3</sub>/TiO<sub>2</sub>/ UiO-66-(COOH)<sub>2</sub>的制备方法及其应用
- 一种石墨烯负载的具有凹面立方体形貌的Ag光催化剂的制备方法
- 一种选择性专一识别的PPyZnFe2O4磁性印迹复合光催化剂的制备方法
- 的制备方法及应用
- 抗病毒性组合物、抗病毒剂、光催化剂及病毒灭活方法
- 一种选择性专一识别的PPyZnFe2O4磁性印迹复合光催化剂的制备方法
- 一种AlON复合可见光催化剂及其制备方法和应用与流程
- 氮化碳光催化材料及其制备方法和应用与流程
- 一种原位制备g‑C3N4‑TiO2纳米异质结光催化薄膜的方法与流程
- 一种银/氯化银复合纳米立方体的制备方法与流程
- 一种BiOCl光催化剂的制备方法及制得的光催化剂和其应用与流程
- 一种BiOCl纳米光催化剂的制备方法及制得的光催化剂和应用与流程
- 一种块状BiOCl光催化剂的制备方法及制得的光催化剂和应用与流程
- 一种CdS量子点/Bi2MoO6复合型光催化剂的制备方法与流程
- 一种纳米层状CdV2O6‑CdS复合光催化剂及其制备方法和应用与流程
- 一种Cd‑Nd‑S/TiO2‑NTs可见光催化材料的制备与应用的制造方法与工艺
- 一种氧化钴修饰的Ag/TiO<sub>2</sub>同轴异质纳米线光催化剂的制备方法
- 二氧化硅纳米片负载铁氮共掺杂TiO<sub>2</sub>光催化剂的方法及用图
- 具有光催化性能的连续纤维素/TiO<sub>2</sub>气凝胶纤维的制备方法
- 一种光致Ti<sup>3+</sup>自掺杂TiO<sub>2</sub>光催化剂的制备方法
- 一种可见光响应的BiVO<sub>4</sub>/TiO<sub>2</sub>/石墨烯三元复合光催化剂及其制备方法
- 一种网载TiO<sub>2</sub>/活性炭的吸附-光催化净水复合材料的制备方法
- TiO<sub>2</sub>/Ga<sub>2</sub>O<sub>3</sub>复合光催化胶体的制备方法
- 一种CoP/ TiO<sub>2</sub>复合光催化剂及其制备和应用
- 一种具有可见光催化的氮掺杂的柔性TiO<sub>2</sub>/SiO<sub>2</sub>纳米纤维薄膜的制备方法
- 一种复合可见光光催化剂Ag<sub>2</sub>CO<sub>3</sub>/TiO<sub>2</sub>/ UIO-66-(COOH)<sub>2</sub>及有机物降解应用