微波辅助‑中空纤维‑液/固微萃取装置及微萃取方法与流程
微波辅助-中空纤维-液/固微萃取装置及微萃取方法技术领域本发明属于化学分析仪器及测试技术领域,特别涉及一种微波辅助-中空纤维-液/固微萃取装置及利用该装置的微萃取方法。
背景技术:
传统样品前处理方法主要有液-液萃取、固相萃取、液-固萃取、索氏抽提、超声辅助萃取、中空纤维-液相微萃取等。中空纤维-液相微萃取技术(HollowFiber-LiquidPhaseMicro-extraction,HF-LPME)是一种新型的样品前处理技术。HF-LPME利用中空纤维的微孔限制、分离无效生物大分子,使其不能进入到萃取溶剂中,从而减少基质干扰,且中空纤维为一次性使用可有效避免交叉污染问题,具有溶剂用量少、净化效率高、操作简便等特点,已广泛应用于环境、生物等复杂样品的分离、萃取。然而,该技术在萃取过程中,依靠分析物以吸附或被动扩散的方式进入萃取溶液,导致平衡时间较长,且目标化合物无法从基体中快速游离出来,仅适用于环境水样的前处理。固相微萃取(solidphasemicroextraction,SPME)是一种集萃取、浓缩、解吸于一体的样品前处理技术,该技术利用目标化合物组分在两相间的分配平衡差异,从而达到使目标化合物与样品基质以及样品中的干扰物分离的效果。固相微萃取技术具有操作简单、无需溶剂、样品分析速度快、样品用量少、重现性好、易于仪器联用等特点,适用于痕量化合物的检测,但固相微萃取装置的萃取头价格昂贵,易磨损,且使用寿命有限,因此,该技术目前还未被广泛应用。在处理复杂基质样品或痕量目标化合物时,这些前处理技术已越来越不能适应现代分离分析的要求,为满足人们对痕量目标化合物分离分析的要求,现有不少学者致力于这方面的研究,国内研发了一种分子印迹固相微萃取-中空纤维液相微萃取联用装置,该装置中的分子印迹固相微萃取探针以石英纤维或金属丝为底材,表面涂覆分子印迹聚合物。然而,该装置采用的是被动扩散的方式达到萃取平衡,耗时较长。国内还研发了一种微波辅助-中空纤维-液/液微萃取装置,该装置虽可有效测得牛奶中的27种抗生素化合物,但存在平行性较差这一问题,这可能是由微波和温度带来的瞬时高温所引起得中空纤维管内有机溶剂的挥发、损失而导致的。
技术实现要素:
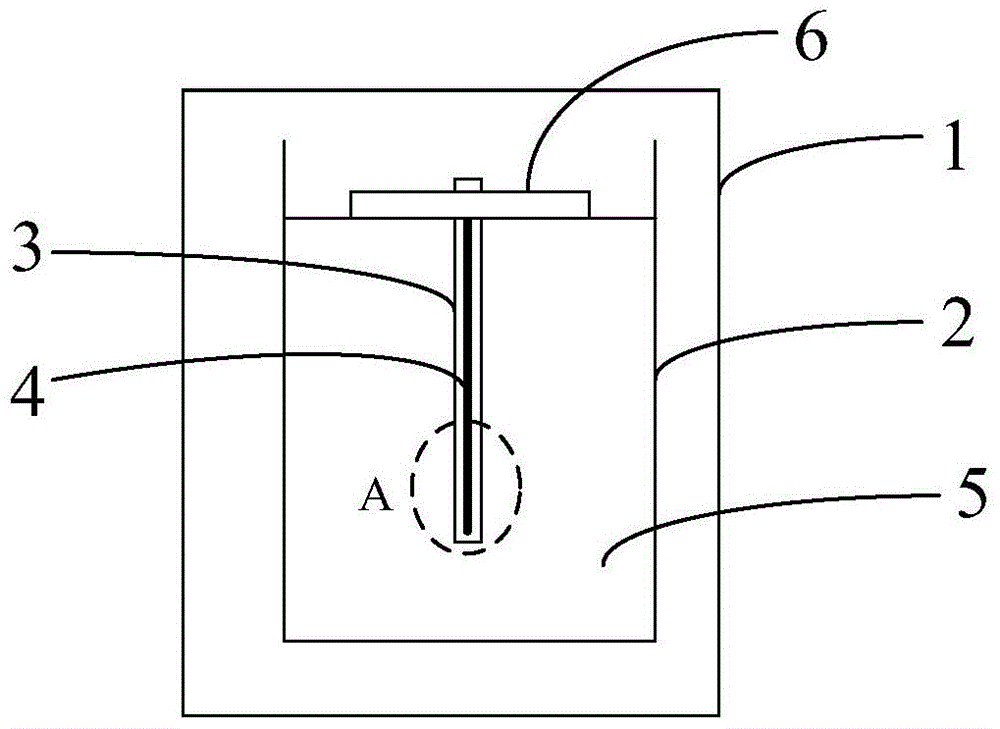
本发明的目的在于克服现有技术的上述不足,提供一种微波辅助-中空纤维-液/固微萃取装置以及利用所述微波辅助-中空纤维-液/固微萃取装置的萃取方法,以克服现有微萃取装置存在的微萃取平衡时间长,成本高和使用寿命短的技术问题。为了实现上述发明目的,本发明的技术方案如下:一种微波辅助-中空纤维-液/固微萃取装置,包括微波腔体和设置在所述微波腔体内的用于盛溶液的容器,在所述容器内设置有两端部密封的中空纤维管,且在所述中空纤维管封闭的腔体内盛装有有机萃取液和设置有固相微萃取纤维,所述固相微萃取纤维没入所述有机萃取液中。以及,利用上述的微波辅助-中空纤维-液/固微萃取装置的微萃取方法,包括如下步骤:将待测样品溶液加入所述容器中,将所述中空纤维没入所述待测样品溶液中后,先采用功率为500-600W的微波对所述测样品溶液加热至50-60℃,然后再采用100-200W的微波对所述测样品溶液微波处理10-20min。上述微波辅助-中空纤维-液/固微萃取装置利用中空纤维的微孔限制,能有效分离无效大分子,使其不能进入有机溶剂萃取相从而减少基质干扰。利用固相微萃取纤维的特性,使其管壁微孔大量吸附萃取相中的目标化合物,从而促进目标化合物持续进入到有机萃取液中,中空纤维的小分子透过性以及液-液、液-固微萃取的双重富集作用可实现对具有复杂基质样品中痕量目标化合物的同步分离、萃取、净化和富集。与此同时,与微波辅助技术联用可有效提高提取效率,加快界面间的传质速率,大大缩短微萃取的平衡时间。本发明装置制作成本低廉,易于操作,不仅有效简化了传统样品前处理的步骤、缩短了微萃取时间,而且还易与后续分析仪器联用。上述利用本发明微波辅助-中空纤维-液/固微萃取装置的萃取方法直接将样品溶液置于容器中即可萃取,样品溶液无需前处理,减少了萃取步骤,且其工艺简单易控,且微萃取时间短,显著的提高了萃取效率。同时,能通过本发明装置中的中空纤维管阻隔,减少基质对固相微萃取纤维的干扰,因此更适合于复杂基体的样品前处理,且适用于复杂基质中多类、痕量目标化合物的分离富集,具有良好的实用价值。附图说明下面将结合附图及实施例对本发明作进一步说明,附图中:图1为本发明实施例微波辅助-中空纤维-液/固微萃取装置的一结构示意图;图2为图1中A部分的放大图;图3为本发明实施例微波辅助-中空纤维-液/固微萃取装置的另一结构示意图;图4为本发明实施例固相微萃取纤维的1000倍SEM图;图5为本发明实施例固相微萃取纤维的5000倍SEM图;图6为利用本发明实施例微波辅助-中空纤维-液/固微萃取装置结合LC-MS/MS对含有抗生素分成的样品进行的平行性(稳定性)分析图;图7利用本发明实施例微波辅助-中空纤维-液/固微萃取装置结合LC-MS/MS对含有抗生素分成的样品进行微萃取和分析结果图。具体实施方式为了使本发明要解决的技术问题、技术方案及有益效果更加清楚明白,以下结合实施例,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明。本发明实施例提供一种微波辅助-中空纤维-液/固微萃取装置,其结构如图1-3所示,该微波辅助-中空纤维-液/固微萃取装置结构包括微波腔体1和容器2,该容器2设置在该微波腔体1内。在一实施例中,如图3所示,该微波辅助-中空纤维-液/固微萃取装置还包括磁力搅拌器8和磁力转子9。其中,该搅拌器8设置在微波腔体1内,且容器2置于磁力搅拌器8上,磁力转子9放置在容器2内。因此,在磁力搅拌器8工作时,该磁力转子9在磁力搅拌器8的磁力作用下在容器2内转动,从而起到对盛装在容器2内的溶液搅拌的作用。该实施例中的磁力搅拌器8的设置可有效增大容器2中样品溶液的传质速率,缩短萃取时间。在图1、图3的实施例中,微波腔体1可以是能提供微波设备的工作间提供,如选用MAS-II微波萃取/反应工作站,当然还可以选用能够提供微波的其他微波设备。该微波腔体1在工作过程中,能够为容器2的样品溶液提供微波处理作用,采用微波辅助技术联用可有效提高目标化合物的提取效率,加快界面间的传质速率,大大缩短微萃取的平衡时间。容器2用于盛装分离萃取的样品溶液,如果可以是含有多类、痕量目标化合物的复杂基质的溶液。该容器2可以选用玻璃容器,如烧杯、烧瓶等,利索当然的还可以是陶瓷等容器,只要是不能屏蔽微波的容器均在本发明公开的范围之内。在该容器2内还内设置有两端部密封的中空纤维管3,且在中空纤维管3封闭的腔体内盛装有有机萃取液7和设置有固相微萃取纤维4,固相微萃取纤维4没入有机萃取液7中,如图1-3,特别是如图2所示。其中,该中空纤维管3具有微孔结构,由于该微孔结构的限制,能有效分离无效大分子如生物大分子,使其不能进入有机溶剂萃取相即有机萃取液7中,从而减少基质干扰。在一实施例中,该中空纤维管3选用管壁孔径为纳米级或微米级的疏水中空纤维管。在另一实施例中,该中空纤维管3选用的内径为0.5mm-3.0mm,具体如1.2mm;壁厚0.5-500μm,具体如1μm、10μm、50μm、100μm、150μm、200μm、300μm、400μm等;管壁所具有的孔隙的孔径为0.05μm-200nm,具体如0.05μm、0.06μm、0.07μm、0.08μm、0.09μm、0.1μm(100nm)、0.15μm、0.2μm。其材质可以是本领域通用的材质,如聚偏氟乙烯。在一实施例中,当中空纤维管3的内径为0.5mm-3.0mm时,该固相微萃取纤维4的直径为0.8-0.5mm。当然,应当理解的是,该固相微萃取纤维4的直径是小于中空纤维管3的内径的。在中空纤维管3在容器2内的方式可以是竖直、水平或斜插等方式放置,在一实施例中,如图1、3所述,在中空纤维管3的一端还套设有以套件6,该套件能够使得中空纤维管3在盛装在容器2的溶液内竖直放置,如图1、3所示。具体的,该套件6可以是离心管的离心盖。有机萃取液7应该是与在盛装容器2内溶液不相溶,通过相似相容原理,溶解在容器2内溶液中的目标化合物如多类、痕量目标化合物通过中空纤维管3管壁的微孔进入中空纤维管3内并溶解在有机萃取液7中。具体地,目标化合物通过中空纤维管3的管壁进入有机萃取液7后,再通过固相微萃取纤维4表面微孔的吸附作用,目标化合物逐渐于固相微萃取纤维4上富集,该富集可打破液-液平衡,促使容器2内的目标化合物源源不断的进入中空纤维管3内的有机液7中。该有机萃取液7可以选用挥发性低,极性小等有机溶剂,在某些实施例中,该有机萃取液7选用正辛醇、甲苯、二甲苯、氯苯、苯中的至少一种。在一具体实施例中,该有机萃取液7为体积比1:1正辛醇、甲苯。该种类的有机萃取液7不仅与中空纤维管3有较强的亲和力,而且对待分析的目标化合物有较好的溶解性。该固相微萃取纤维4是具有多孔结构的,在一实施例中,该固相微萃取纤维4所具有的孔隙孔径为0.1~20μm。在一实施例中,该固相微萃取纤维4可以按照如下方法制备获得:S01.配制含α-甲基丙烯酸、苯乙烯、有机交联剂、有机致孔剂、偶氮引发剂混合有机溶液:将α-甲基丙烯酸、苯乙烯、有机交联剂、有机致孔剂、偶氮引发剂进行混料处理,得到混合有机溶液;S02:将所述混合有机溶液置于无氧环境中后,向步骤S01配制的混合有机溶液中投放毛细管,并使得所述混合有机溶液填充至所述毛细管的腔体内;S03:继续在无氧环境中,将投放有所述毛细管的所述混合有机溶液加热至50-70℃进行聚合反应,待所述聚合反应结束后对所述毛细管洗涤、老化处理和除去所述毛细管获得固相微萃取纤维4。具体地,上述步骤S01中,α-甲基丙烯酸、苯乙烯作为制备固相微萃取纤维4的聚合单体。在一实施例中,该两单体的混合比例可以按照α-甲基丙烯酸、苯乙烯的体积比为(3-4):1,在具体实施例中,α-甲基丙烯酸、苯乙烯的体积比可以是为3.0:1、3.2:1、3.5:1、3.7:1、3.9、4:1等。控制单体α-甲基丙烯酸、苯乙烯的比例,使得在α-甲基丙烯酸聚合成水溶性聚合物的过程中,加入该比例的苯乙烯可有效提高聚合产物的亲脂性能。此外,该比例的苯乙烯对聚合产物的机械性能有利,且苯乙烯可提高交联密度,使其能更好的阻止化学试剂的“进攻”。在发明人研究中发现,当随着苯乙烯加入量增大时,如大于1时,会导致聚合产物的亲水性能降低。上述有机交联剂能使得α-甲基丙烯酸、苯乙烯单体在聚合反应过程中相互键合交联成网状结构物质,也即是即固相微萃取纤维4,为了使得α-甲基丙烯酸、苯乙烯单体交联反应所形成的网状结构物质性能更加稳定,在一实施例中,该有机交联剂的加入的量与所述α-甲基丙烯酸的体积比为(2-3.5):1;在以具体实施例中,该有机交联剂与α-甲基丙烯酸的体积比2.0:1、2.1:1、2.2:1、2.3:1、2.5:1、2.8:1、3:1、3.2:1、3.4:1、3.5:1等。该比例的致孔剂能够使得聚合产物的交联度越高,比表面积越大,从而提高聚合产物的吸附能力、萃取容量和富集倍数。在另一优选实施例中,该有机交联剂选自二甲基丙烯酸乙二醇、三甲基丙烷三甲基丙烯酸酯中的一种或两者复合物。上述有机致孔剂在α-甲基丙烯酸、苯乙烯单体形成网状结构固相微萃取纤维4过程中,使得固相微萃取纤维4存在适合的孔隙,从而增大该固相微萃取纤维4的表面积,进而增大固相微萃取纤维4的其富集能力,提高萃取容量。因此,在一实施例中,该有机致孔剂选自甲苯、邻苯二甲酸二甲脂中的一种或两者复合物。在另一实施例中,该有机致孔剂的加入的量与所述α-甲基丙烯酸的体积比为(3-4):2;在以具体实施例中,该有机致孔剂与α-甲基丙烯酸的体积比3.0:2、3.2:2、3.4:2、3.5:2、3.7:2、3.8:2、3.9:2、4.0:2。上述偶氮引发剂能在50-70℃下只形成一种自由基,且分解均匀,为一级反应,不发生诱导分解,无其他副反应,从而使得α-甲基丙烯酸与苯乙烯两单体间进行的聚合反应稳定,反应条件较易控制。而且在聚合反应过程中,该偶氮类引发剂对溶剂和杂质不敏感,通过引入极性基团可增加聚合产物的水溶性(一些杂质的存在易在聚合中引起链转移反应,且易使引发过程重现性变差,导致产品质量不稳定,而偶氮类引发剂对杂质不敏感,因此可有效避免上述情况的发生)因此,在一实施例中,该偶氮引发剂选自偶氮二异丁腈、偶氮二异庚腈和偶氮二异丁酸二甲酯中的至少一种。具体的,该偶氮引发剂可以选用偶氮二异丁腈、偶氮二异庚腈和偶氮二异丁酸二甲酯中的任意一种;也可以是偶氮二异丁腈与偶氮二异庚腈的复合物、偶氮二异庚腈与偶氮二异丁酸二甲酯的复合物、偶氮二异丁腈与偶氮二异丁酸二甲酯的复合物,此时,在下述实施例中,偶氮二异庚腈与偶氮二异丁腈的质量比为1.5:1;偶氮二异庚腈与偶氮二异丁酸二甲酯的质量比为1.1:1;偶氮二异丁酸二甲酯与偶氮二异丁腈的质量比为1.4:1。当然还可以是偶氮二异丁腈、偶氮二异庚腈和偶氮二异丁酸二甲酯三者的复合物,此时,偶氮二异丁腈、偶氮二异庚腈和偶氮二异丁酸二甲酯之间的质量用量比可以是0.73:1.1:1。上述关于偶氮引发剂的各实施例中,其用量可以是足量。在一实施例中,该偶氮引发剂的添加量与α-甲基丙烯酸的质量比为(14-15):1。在具体实施例中,偶氮引发剂与α-甲基丙烯酸的质量比为14.1:1、14.3:1、14.5:1、14.7:1、14.8:1、15:1等。另外,上述步骤S01中,并没有添加其他溶剂,而是利用α-甲基丙烯酸、苯乙烯、有机交联剂、有机致孔剂、偶氮引发剂等自身的溶剂特性,使得各反应充分互相混合和溶解,形成均匀的混合有机溶液。因此,对于该步骤S01配制的混合有机溶液中,是绝对比容许水等无机溶剂的存在的。为了使得该步骤S01各反应物充分的混合和溶解,在一实施例中,将各反应物混合后,可以将混合液采用超声处理,具体的可以采用超声震荡处理15分钟。上述步骤S02中,无氧环境是为了步骤S01中配制的使得混合有机溶液进行聚合反应提供一无氧的环境,保证反应物如偶氮引发剂失去活性。该无氧环境可以是真空环境或惰性的气体环境。具体地,真空环境可以采用抽真空设备来实现,惰性的气体环境可以是充满氮气、氩气等保护性气氛的环境。向该混合有机溶液投放的毛细管是否可以根据实际的需要进行选择规格,在研究中发现,毛细管内径对制备的固相微萃取纤维4的萃取结果基本没有影响。具体的可能根据中空纤维管3的空腔直径大小而选用。因此,该毛细管可以选用25μL毛细点样管规格:长:125mm,外径/内经。上述步骤S03中,无氧环境如同上述步骤S02的无氧环境。在无氧环境中将混合有机溶液加热至50-70℃并保持该温度直至混合有机溶液反应完毕。在该50-70℃下,反应单体α-甲基丙烯酸与苯乙烯在偶氮引发剂、有机交联剂等作用下发生聚合反应,在此过程中有机致孔剂参与了反应,与其它有机试剂相互交联聚合形成多空骨架结构。为了使得混合有机溶液反应稳定,在毛细管腔体中形成的固相微萃取纤维4更加符合本发明实施例的要求,在一实施例中,该聚合反应的温度为60℃。在另一实施例中,对混合有机溶液升温的速率是20±2℃/分钟上述聚合反应的时间应该保证聚合反应充分反应直至反应完毕,如在一实施例中,反应时间可以控制在4-8小时,在一具体实施例中,该聚合反应在60℃下持续反应6小时。该步骤S03中,待聚合反应完毕,将毛细管取出对其清洗,以除去毛细管表面过量的反应物以及其他副产物等杂质。清洗方式可以采用无水乙醇进行清洗,在清楚过程中还可以伴随超声处理。除去毛细管的方式可以采用破坏毛细管的方式除去,从而获得在毛细管腔体内形成的固相微萃取纤维4。当然还可以用其他能除去毛细管的方法以获得腔体内形成的固相微萃取纤维4。为了进一步提高固相微萃取纤维4的机械性能,在进一步实施例中,在除去毛细管的处理之前,对所述毛细管洗涤处理之后,还包括对毛细管腔体内的固相微萃取纤维4进行老化处理。在一实施例中,该老化处理可以按照如下方法进行:将清洗干燥后的毛细管于110-150℃下热处理6-8小时后冷却。在一些具体实施例中,该老化为110℃、115℃、120℃、125℃、130℃、135℃、140℃、145℃、150℃。在一具体实施例中,将清洗干燥后的毛细管于于120℃下热处理7小时后冷却。应当理解的是,经除去毛细管处理后,还包括将获得的固相微萃取纤维4进行清洗处理的步骤,该清洗处理的方法可以是:用无水乙醇和甲醇分别将固相微萃取纤维4进行超声清洗,如可以清洗十遍。上述固相微萃取纤维制备方法以反应物自身提供有机相的反应体系,采用偶氮化合物作为单体聚合反应的引发剂,使得聚合反应反应稳定,无副反应产物少,并使得该聚合反应条件要求低,易控制,有效提供了生产效率,降低了生产成本。由该固相微萃取纤维制备方法制备得到固相微萃取纤维4表面光滑,外观呈均匀乳白色,在扫描电子显微镜下,该纤维表面粗糙,具有较均匀的孔状结构,具体如图4、5所示。由此,该固相微萃取纤维4的性能稳定,能适用于多种亲水/亲脂型化化合物的富集,且其吸附能力强,萃取容量大,富集倍数高。在此基础上,能有效的提高上述微波辅助-中空纤维-液/固微萃取装置的微萃取效果。在上述微波辅助-中空纤维-液/固微萃取装置实施例的基础上,本发明还提供了利用上述的微波辅助-中空纤维-液/固微萃取装置的微萃取方法的实施例。该利用微波辅助-中空纤维-液/固微萃取装置的萃取方法包括如下步骤:将待测样品溶液加入微波辅助-中空纤维-液/固微萃取装置的容器2中,将中空纤维3置于容器2内并没入所述待测样品溶液中后,利用微波腔体1提供的微波,先采用功率为500-600W的微波对所述测样品溶液加热至50-60℃,然后再采用100-200W的微波对所述测样品溶液微波处理10-20min。利用微波辅助-中空纤维-液/固微萃取装置在该萃取的工艺和条件能有效缩短微萃取时间,显著的提高了萃取效率。为了使得微萃取时间更短,效率更高,在一实施例中,对该盛装在容器2内待测样品溶液微萃取的工艺和条件:先采用功率为600W的微波对所述测样品溶液加热至60℃,然后再采用200W的微波对所述测样品溶液微波处理19min。上述微萃取方法实施例中,待测样品可以是含有多类、痕量目标化合物的复杂基质。如水样、土壤、尿液、生物样品提取液、饮料、牛奶中的任一种。其中,土壤可以是沉积泥。上述微萃取方法实施例中,理所当然的是,在将待测样品溶液盛装在容器2中之前或之后还包括对中空纤维管3的处理和制备。对中空纤维管3的处理和制备方法如下:(1)将中空纤维管3清洗晾干处理后对其一端进行封口处理;(2)将固相微萃取纤维4置于步骤(1)中空纤维管3内;(3)于步骤(2)中空纤维管3内注入有机萃取液7;(4)将步骤(3)中空纤维管3另一端封口后置于盛装有测样品溶液的容器2内。其中,步骤(1)中中空纤维管3清洗处理可以是采用有机溶剂如甲醇的水溶液其清洗处理,具体地,甲醇的水溶液中,甲醇与水的体积比可以是1:1;清洗处理可以是超声清洗处理。对清洗处理后的中空纤维管3封口处理可以直接采用封口机进行。步骤(2)中,在将固相微萃取纤维4之前,优选的还包括将中空纤维管3置于有机萃取液7中进行浸泡处理,使得该有机萃取液7填充至中空纤维管3管壁的孔隙中。现以微波辅助-中空纤维-液/固微萃取装置和利用微波辅助-中空纤维-液/固微萃取装置进行的微萃取方法为例,对本发明进行进一步详细说明。实施例1一种微波辅助-中空纤维-液/固微萃取装置,其结构如图3所示。该装置中的固相微萃取纤维4由如下方法制备获得:S11:准确量取672μLα-甲基丙烯酸、168μL苯乙烯、2016μL二甲基丙烯酸乙二醇酯、1008μL甲苯、45.6mg偶氮二异丁腈;S12:将步骤S11中量取的各组分在常温下超声振荡15min,使得各成分混合均匀并溶解;S13:将步骤S12中配制的混合溶液移至表面皿中,并用保鲜膜密封(仅留一个小孔),吹氮5min;S14:将规格为50μL(规格:长:125mm,外径/内经)的毛细管投入表面皿中,使毛细管内充满混合溶液,继续吹氮2min;S15:用保鲜膜将表面皿完全密封,并于60℃烘箱内聚合反应6h;S16:用无水乙醇超声清洗四遍,充分洗去毛细管外壁残留液,于通风橱内晾干;S17:于120℃烘箱内老化处理7h;S18:将经步骤S17老化处理的毛细管壁敲碎,先用乙醇超声清洗10遍,再用甲醇超声清洗5遍,将合成出的固相微萃取纤维(SPME)萃取纤维超声清洗十遍。将本实施例1制备的固相微萃取纤维采用SEM分别在1000倍和5000倍进行分析,1000倍分析结果如图4所示,5000倍分析结果如图5所示。实施例2一种微波辅助-中空纤维-液/固微萃取装置,其结构如图3所示。其中,该装置中的固相微萃取纤维4由如下方法制备获得:S21:准确量取504μLα-甲基丙烯酸、168μL苯乙烯、1008μL三甲基丙烷三甲基丙烯酸酯、2016μL邻苯二甲酸二甲脂、34.95mg偶氮二异庚腈;S22:将步骤S21中量取的各组分在常温下超声振荡20min,使得各成分混合均匀并溶解;S23:将步骤S22中配制的混合溶液移至表面皿中,并用保鲜膜密封(仅留一个小孔),吹氮8min;S24:将规格为25μL(规格:长:125mm,外径/内经)的毛细管投入表面皿中,使毛细管内充满混合溶液,继续吹氮10min;S25:用保鲜膜将表面皿完全密封,并于50℃烘箱内聚合反应8h;S26:用无水乙醇超声清洗四遍,充分洗去毛细管外壁残留液,于通风橱内晾干;S27:于150℃烘箱内老化处理6h;S28:将经步骤S27老化处理的毛细管壁敲碎,先用乙醇超声清洗10遍,再用甲醇超声清洗5遍,将合成出的固相微萃取纤维(SPME)萃取纤维超声清洗十遍。实施例3一种微波辅助-中空纤维-液/固微萃取装置,其结构如图3所示。其中,该装置中的固相微萃取纤维4由如下方法制备获得:S31:准确量取588μLα-甲基丙烯酸、168μL苯乙烯、1008μL二甲基丙烯酸乙二醇酯、1008μL三甲基丙烷三甲基丙烯酸酯、336μL甲苯、676μL甲苯邻苯二甲酸二甲脂、399mg偶氮二异丁酸二甲酯;S32:将步骤S31中量取的各组分在常温下超声振荡10min,使得各成分混合均匀并溶解;S33:将步骤S32中配制的混合溶液移至表面皿中,并用保鲜膜密封(仅留一个小孔),吹氮10min;S34:将规格为50μL(规格:长:125mm,外径/内经)的毛细管投入表面皿中,使毛细管内充满混合溶液,继续吹氮5min;S35:用保鲜膜将表面皿完全密封,并于70℃烘箱内聚合反应4h;S36:用无水乙醇超声清洗四遍,充分洗去毛细管外壁残留液,于通风橱内晾干;S37:于110℃烘箱内老化处理8h;S38:将经步骤S37老化处理的毛细管壁敲碎,先用乙醇超声清洗10遍,再用甲醇超声清洗5遍,将合成出的固相微萃取纤维(SPME)萃取纤维超声清洗十遍。实施例4一种微波辅助-中空纤维-液/固微萃取装置,其结构如图3所示。其中,该装置中的固相微萃取纤维4由如下方法制备获得:该固相微萃取纤维4的制备方法参照实施例1中固相微萃取纤维4的制备方法,不同之处在于偶氮二异丁腈采用质量比为1:0.73的偶氮二异丁腈与偶氮二异丁酸二甲酯的复合物替代。实施例5一种微波辅助-中空纤维-液/固微萃取装置,其结构如图1所示。S51:准确量取672μLα-甲基丙烯酸、168μL苯乙烯、2016μL二甲基丙烯酸乙二醇酯、1008μL甲苯、45.6mg偶氮二异丁腈;S52:将步骤S51中量取的各组分在常温下超声振荡15min,使得各成分混合均匀并溶解;S53:将步骤S52中配制的混合溶液移至表面皿中,并用保鲜膜密封(仅留一个小孔),吹氮5min;S54:将规格为50μL(规格:长:125mm,外径/内经)的毛细管投入表面皿中,使毛细管内充满混合溶液,继续吹氮2min;S55:用保鲜膜将表面皿完全密封,并于60℃烘箱内聚合反应6h;S56:用无水乙醇超声清洗四遍,充分洗去毛细管外壁残留液,于通风橱内晾干;S57:将经步骤S56老化处理的毛细管壁敲碎,先用乙醇超声清洗10遍,再用甲醇超声清洗5遍,将合成出的固相微萃取纤维超声清洗十遍。实施例6利用实施例1中提供的微波辅助-中空纤维-液/固微萃取装置进行微萃取并结合LC-MS/MS对水样中的喹诺酮类、磺胺类、镇静剂、消炎镇痛类、香料类、大环内酯类等共55种抗生素化合物进行测定。方法如下:将中空纤维管3统一裁至适合长度(约9cm),经体积比为1:1的甲醇/水超声清洗15min后,再用丙酮溶液超声清洗5min,晾干,用封口机对其一端进行热封口,将其浸入正辛醇/甲苯(v:v=1:1)溶液中超声10min,使中空纤维管3壁的孔隙中形成一种有机溶剂膜。用微量注射器将中空纤维管3腔内的多余溶剂吸出,仅使中空纤维管3管壁及微孔充满溶剂,将固相微萃取纤维4插入中空纤维管3内,用微量注射器向中空纤维管3内注入50μL的正辛醇/甲苯溶液(v:v=1:1),并将中空纤维管3的另一端热封口。将中空纤维管3一端固定于50ml离心管盖6上,使装有溶剂的中空纤维管3全部浸没于供相溶液中(供相溶液含18.75g氯化钠、100ml自来水和2μg/L的55种抗生素药物,该混标为55种单标药物混合配置而成),再将样品瓶置于MAS-II微波萃取/反应工作站微波腔体1中,在700r/min的搅拌速度下,先于600W微波功率下微波、搅拌60s,将溶液加热至60℃,再于100W微波功率下反应10min,且保持反应温度为60℃,取出SPME萃取纤维并置于5ml离心管内,加入8ml色谱甲醇,超声振荡15min,于氮气浓缩仪中吹氮浓缩并定容至0.5ml,供LC-MS/MS测定。性能测试(一)将上述实施例1-5制备的固相微萃取纤维进行如表1中性能测试,各项性能测试结果如下表1所述:表1由表1可知,本发明实施例制备的固相微萃取纤维4分布均匀的孔隙,比表面积大,具有优异的亲水/亲脂化化合物的富集性能、优异的稳定性能。另外,通过测试,其还具有良好的机械强度。(二)将实施例6微萃取进行平行性(稳定性)分析和LC-MS/MS测定,其结果如下:1.微波辅助-中空纤维-液/固微萃取装置结合LC-MS/MS对含有抗生素分成的样品进行平行性分析结果如图6所述,图6中并排的3根柱状图表示的是对同一目标待测物的三组平行实验结果,依次是平行组1、平行组2、平行组3。通过三组平行空白水样萃取结果对比可知,本发明实施例提供的微波辅助-中空纤维-液/固微萃取装置对目标化合物微萃取稳定性好。且说明了本发明实施例提供的微波辅助-中空纤维-液/固微萃取装置有效克服了现有微波辅助-中空纤维-液/液微萃取法因微波和温度带来的瞬时高温所引起得中空纤维管内有机溶剂的挥发、损失从而导致的平行性差的不足。此外,该装置还具有操作简单、溶剂用量更少、萃取时间更短、稳定性更好等特点。2.微波辅助-中空纤维-液/固微萃取装置结合LC-MS/MS测定结果如图7所示。由图7a至图7z说明了本发明实施例提供的微波辅助-中空纤维-液/固微萃取装置结合LC-MS/MS能对目标样品进行精确的定量分析。以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1