一种具有极高选择性的测定锌电解液中Co2+含量的络合物吸附波极谱法的制作方法
本发明涉及一种直接检测锌电解液中微量或痕量Co2+的络合物吸附波极谱法。
背景技术:
锌是支撑国民经济和国防军工发展的基础原料和战略性物资,目前80%左右的锌是通过湿法冶金产生的。湿法炼锌工艺依次主要包括磨矿、浸出、净化和电解过程。其中净化工序是用来控制电解液中杂质离子浓度的关键工序,通过待净化液中杂质的种类和含量来判断需要加入的除杂试剂的种类和用量。目前大部分冶炼厂仍采用手工间歇性的传统方法来分析有关杂质的种类和含量,这些方法过程繁琐、耗时,导致检测信息反馈滞后时间长。这使得净化工序的工艺参数无法实现实时优化调整,往往须采用过量加入锌粉以保证除杂效果,导致净化后料液杂质离子浓度波动大、锌粉大量浪费、生产成本高等问题。因此,开发检测步骤简单、速度快,适用于在线检测的方法至关重要。在电解过程中,杂质金属离子在阴极放电析出严重地影响析出锌的结晶状态、电积过程的电流效率和电解锌的质量。钴是湿法炼锌浸出液中最难除去的杂质之一,且对锌的电沉积危害非常大,当其浓度超过一定值时,就会引起“烧板”现象和降低电流效率等危害,因此需要严格监控锌电解液中钴的含量。
微量Co2+的测定方法很多,主要有分光光度法,原子吸收法,原子发射光谱法,极谱法,色谱法,质谱法等。由于锌电解液中Zn2+的含量很高(130~170g/L),锌钴的质量比最高可达到107倍以上,以至严重干扰锌电解液中Co2+的测定,这些方法都不能满足简便快速实现钴测定的要求。目前部分冶炼厂采用亚硝基R盐吸光光度法来测定Co2+,但方法灵敏度低,线性范围较窄(0.3~5μg·mL-1),且须反复多次加热煮沸,分析速度慢,难以快速准确地得到分析结果,不能在线监测除杂效果和净化液质量。所以寻找新的测试方法至关重要。
络合物吸附波极谱法一般灵敏度高,选择性好,仪器简单,分析速度快,非常适合微量或痕量元素的测定和在线分析检测仪器的研究开发,目前关于络合物吸附波测定水溶液中的Co2+已有报道,而且大部分方法采用中性或碱性(多为氨性)介质,Co2+的峰电位都在-1.0V(vs.SCE)左右,与Zn2+的峰电位很接近,因而受Zn2+的干扰相当严重,当用来测定锌电解液中含量较低的钴时Co2+波则完全被Zn2+波掩盖而无法测定,若采用预处理方法沉淀Zn2+,不仅操作麻烦,而且在在线分析中,沉淀的生成容易堵塞管路,不易清理。
技术实现要素:
本发明的目的是针对锌电解液中Co2+的测定,提供一种简单,快捷,高选择性和高灵敏度的能够用于在线分析的络合物吸附波极谱检测方法。
本发明通过以下方案来实现:
一种具有极高选择性的测定锌电解液中Co2+含量的络合物吸附波极谱法,包括将待测样品锌电解液与检测体系反应,测定反应混合物中产生的高选择性和灵敏度的Co2+络合物吸附极谱波,获得二阶导数波峰电流,从而计算待测样品锌电解液中的Co2+浓度;所述检测体系包括底液氨-氯化铵缓冲液,掩蔽剂乙二胺四乙酸盐,络合剂丁二酮肟或者络合剂丁二酮肟和亚硝酸钠。
本发明方法检测体系选取乙二胺四乙酸盐作为锌电解液中基体成分Zn2+和其他金属离子的掩蔽剂,丁二酮肟、或丁二酮肟和亚硝酸钠作为待测离子Co2+的络合剂(或混配络合剂),其中丁二酮肟作为测Co2+的主络合剂、亚硝酸钠作为辅助络合增敏剂,以氨-氯化铵缓冲液为底液(介质),将待测样品锌电解液与检测体系反应,测定反应混合物中产生的高选择性和灵敏度的Co2+络合物吸附极谱波,锌和电解液中其它离子对Co2+检测都不产生干扰。在常温下测定锌电解液中微量或痕量Co2+时,该方法不需预处理可直接用示波极谱仪等仪器测定。本发明不仅可以完全掩蔽Zn2+,消除电解液中高浓度基体成分Zn2+(锌钴质量比≥107)对Co2+测定的影响,而且可完全掩蔽锌电解液中其他共存杂志离子的干扰,是定性和定量测定锌电解液中微量或痕量钴的高选择性和灵敏的方法。
优选地,所述反应混合物中乙二胺四乙酸盐的摩尔浓度与锌电解液中Zn2+摩尔浓度的比值为1~5,进一步优选为1.1。
优选地,所述乙二胺四乙酸盐为乙二胺四乙酸二钠,具体可用EDTA的氢氧化钠溶液,所述反应混合物中EDTA的浓度为0.005~0.007mol/L,优选为0.006mol/L。
优选地,所述EDTA的氢氧化钠溶液中EDTA的浓度为0.25~0.35mol/L,进一步优选为0.30mol/L。
优选地,所述反应混合物中氨-氯化铵的摩尔浓度为0.3~0.6mol/L,优选为0.48mol/L。
优选地,所述氨-氯化铵缓冲液的浓度为2.0-4.0mol/L,优选为3.0mol/L,其中NH3与NH4Cl的摩尔比为1:9。
当待测样品锌电解液中Co2+浓度为1.0×10-10~4.0×10-4mol/L,优选为1.2×10-9~4.0×10-4mol/L时,所述络合剂为丁二酮肟.
当待测样品锌电解液中Co2+浓度为2.0×10-12~3.2×10-6mol/L,优选为5.0×10-11~3.2×10-6mol/L时;所述络合剂为丁二酮肟和亚硝酸钠。
优选地,所述反应混合物中丁二酮肟的摩尔浓度为7.6×10-4~3.8×10-3mol/L,优选为2.7×10-3mol/L;所述反应混合物中亚硝酸钠的摩尔浓度为0.16~0.48mol/L,优选为0.32mol/L。
优选地,所述丁二酮肟以丁二酮肟的乙醇溶液的形式添加,其浓度为5~12g/L,进一步优选为11g/L。
优选地,所述亚硝酸钠以亚硝酸钠溶液的形式添加,其浓度为2.0~6.0mol/L,进一步优选为4.0mol/L。
本发明方法不需预处理可直接可采用示波极谱仪等仪器进行测定,例如采用JP-303E型示波极谱仪。
测定条件包括:
起始电压:-100~-1050mV,优选为-900mV;
终止电压:-1250~-1500mV,优选为-1300mV;
扫描速率:100~500mV/s,优选为300mV/s;
静止时间:5~14s,优选为6s;
滴汞周期:5~18s,优选为9s。
当电解新液、电解废液、除钴后液等待测样品锌电解液中Co2+浓度较低(例如痕量)时,所述络合剂为丁二酮肟和亚硝酸,即采用丁二酮肟-EDTA-亚硝酸钠-“氨-氯化铵缓冲液”体系(简称体系一)测定Co2+,Co2+线性范围为:5.0×10-11~3.2×10-6mol/L(见图1),相关系数:0.9997,检出限:2.0×10-12mol/L。具体地,所述痕量指锌电解液中Co2+浓度为2.0×10-12~3.2×10-6mol/L,优选为5.0×10-11~3.2×10-6mol/L。
当待测样品锌电解液中Co2+浓度较高(例如微量)时,可适当稀释后再采用上述“体系一”进行测定。
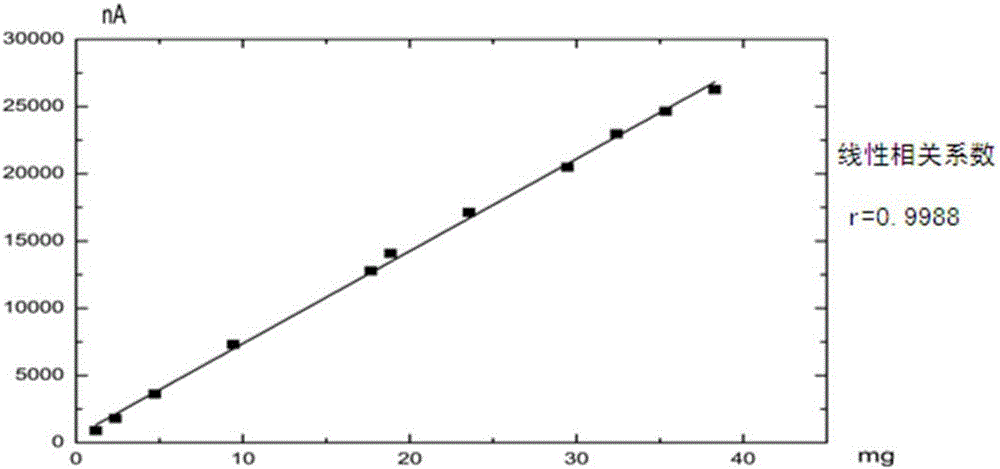
为避免反复稀释的麻烦,当中性上清液等待测样品锌电解液中Co2+浓度较高(例如微量)时,所述络合剂为丁二酮肟,即采用丁二酮肟-EDTA-“氨-氯化铵缓冲液”体系(简称体系二)测定Co2+,即省略加入亚硝酸钠的步骤;Co2+线性范围为:1.2×10-9~4.0×10-4mol/L(见图2),相关系数:0.9988,检出限:1.0×10-10mol/L。具体地,所述微量指锌电解液中Co2+浓度为1.0×10-10~4.0×10-4mol/L,优选为1.2×10-9~4.0×10-4mol/L。
具体地,上述测定方法包括以下步骤:
1)将一定量的待测样品锌电解液与一定量的所述检测体系反应,测定反应混合物中产生的Co2+络合物吸附极谱波(采用示波极谱仪摄图,下同),获得二阶导数波峰电流I1;
2)分别取与步骤1)等量的待测样品锌电解液、与步骤1)等量的所述检测体系及一定量的浓度为a mol/L的Co2+标准溶液,将三者反应测定反应混合物中产生的Co2+络合物吸附极谱波,获得二阶导数波峰电流I2;
3)按下式计算待测样品锌电解液中的Co2+浓度,单位mol/L:
a I1/(I2–I1);
优选地,a为5.0×10-11-4.0×10-4。
更具体地,上述测定方法包括以下步骤:
1)量取0.10mL的待测样品锌电解液于50mL容量瓶中,加入丁二酮肟的乙醇溶液1.4mL,晃动1min,然后依次加入EDTA的氢氧化钠溶液1.0mL,氨-氯化铵缓冲液8.0mL,或者进一步再加入亚硝酸钠溶液4.0mL,静止5min,把所得混合溶液倒入电解杯中测定产生的Co2+络合物吸附极谱波(采用示波极谱仪摄图,下同),获得二阶导数波峰电流I1;
2)量取0.10mL浓度为a mol/L的Co2+标准溶液和0.10mL的待测样品锌电解液于50mL容量瓶中,加入丁二酮肟的乙醇溶液1.4mL,晃动1min,然后依次加入EDTA的氢氧化钠溶液1.0mL,氨-氯化铵缓冲液8.0mL,或者进一步再加入亚硝酸钠溶液4.0mL,静止5min,把所得混合溶液倒入电解杯中测定产生的Co2+络合物吸附极谱波(采用示波极谱仪摄图,下同),获得二阶导数波峰电流I2;
3)按下式计算待测样品锌电解液中的Co2+浓度,单位mol/L:
a I1/(I2–I1);
其中,
丁二酮肟的乙醇溶液浓度为11g/L;
EDTA的氢氧化钠溶液中EDTA的浓度为0.30mol/L;
氨-氯化铵缓冲液的浓度为3.0mol/L,其中NH3与NH4Cl的摩尔比为1:9;
亚硝酸钠溶液浓度为4.0mol/L;
a为5.0×10-11-4.0×10-4;
当所述待测样品锌电解液中Co2+浓度为2.0×10-12~3.2×10-6mol/L,优选为5.0×10-11~3.2×10-6mol/L时;所述络合剂为丁二酮肟和亚硝酸钠;
当待测样品锌电解液中Co2+浓度为1.0×10-10~4.0×10-4mol/L,优选为1.2×10-9~4.0×10-4mol/L时,所述络合剂为丁二酮肟。
步骤1)和步骤2)测定Co2+络合物吸附极谱波的条件相同,具体包括:
仪器:JP-303E型示波极谱仪;
起始电压:-900mV;
终止电压:-1300mV;
扫描速率:300mV/s;
静止时间:6s;
滴汞周期:9s。
当所述待测样品锌电解液中含有微量的Co2+,优选Co2+浓度≥3.2×10-6mol/L时,可将待测样品锌电解液适当稀释后再进行测定,例如稀释10-1000倍。
本发明所述待测样品包括锌电解液、锌电解新液、中性上清液等。
上述两体系中对Co2+测定相对误差不超过5%。下列质量倍数的离子:Zn2+(4×107)、W(VI)和Mn2+(3×104)、Mo(VI)和Hg2+(1×104)、Pb2+(3×103)、Cu2+和Cd2+(1000)、Sb3+(500)、Ni2+(450)、As3+(110)、Fe3+(20)及常见阴离子均不干扰测定。
用上述两个体系直接测定锌电解液中钴,锌电解液中其它共存离子对Co2+的测定均无干扰,满足测定要求。
本发明的思路是要求分析方法操作简单,选择性高,灵敏度高,分析时间短,不需要对样品进行预处理,仪器及试剂价格便宜,不产生沉淀,能够用于在线分析。氨羧络合剂具有广泛而强大的络合能力,与多种金属离子形成的络合物很稳定,且能溶于水。乙二胺四乙酸盐(EDTA)作为应用最广泛的氨羧络合剂,性质稳定,价格便宜,能与大部分金属离子形成配位比为1:1的稳定螯合物。实验发现,Co2+与丁二酮肟形成的络合物的稳定常数非常大,甚至远超过Co2+与EDTA形成络合物的稳定常数,而其它金属离子与丁二酮肟生成的络合物其稳定性则小的多。在丁二酮肟存在下,EDTA与锌电解液中除Co2+之外所有金属离子发生螯合反应,且在测定钴的条件下其螯合物不产生络合物吸附波,因而所有与Co2+共存的金属离子都被掩蔽,而Co2+与丁二酮肟(或丁二酮肟及亚硝酸钠)形成的络合物在氨-氯化铵底液中则产生灵敏的络合物吸附波。因此,此方法对于锌电解液中Co2+的测定具有非常好的选择性和灵敏度。
本发明采用EDTA作为基体成分锌和共存杂质金属离子的掩蔽剂,为了避免因测定较高含量钴须联结稀释装置使在线检测仪器结构复杂化,可采用两个体系分别对微量和痕量Co2+进行测定。在痕量时采用丁二酮肟-EDTA-亚硝酸钠-氨缓冲体系测定Co2+,在微量时采用丁二酮肟-EDTA-氨缓冲体系测定Co2+,试剂简单方便配制,大大提高了自动化程度。本发明方法不仅可以完全掩蔽Zn2+,消除Zn2+波对Co2+测定的影响,而且也可掩蔽锌电解液中其他共存杂质金属离子的干扰,选择性极好,不需要对锌电解液预处理,没有沉淀生成,分析速度快,适合在线分析检测使用。
本发明与现有方法相比,具有如下有益效果:
1.无需对被测锌电解液进行预处理;
2.线性范围宽,对整个工艺流程的Co2+都可以检测;
3.所用仪器价格便宜,操作简单。所用试剂种类少,稳定性好,价格便宜;
4.分析速度快;
5.无沉淀生成;不仅适用于实验室中常规分析,更易实现自动化,便于在线检测中使用。
附图说明
图1为实施例1痕量(较低浓度)钴测定体系的线性回归曲线;
图2为实施例2微量(较高浓度)钴测定体系的线性回归曲线;
图3为实施例3(采用体系一)测定锌电解新液中Co2+的二阶导数极谱波;
图4为实施例4(采用体系二)测定中性上清液中Co2+的二阶导数极谱波;
图5为实施例5(采用体系一)测定中性上清液中Co2+的二阶导数极谱波。
具体实施方式
以下实施例用于说明本发明,但不用来限制本发明的范围。实施例中未注明具体技术或条件者,按照本领域内的文献所描述的技术或条件,或者按照产品说明书进行。所用试剂或仪器未注明生产厂商者,均为可通过正规渠道商购买得到的常规产品。
以下实施例所用试剂及规格:
丁二酮肟的乙醇溶液:11g/L;EDTA的氢氧化钠溶液中EDTA的浓度:0.30mol/L;氨-氯化铵缓冲液:3.0mol/L;NH3与NH4Cl的摩尔比为1:9;亚硝酸钠溶液:4.0mol/L。
以下实施例测试条件:
仪器:JP-303E型示波极谱仪;起始电压:-900mV;终止电压:-1300mV;扫描速率:300mV/s;静止时间:6s;滴汞周期:9s。
以下实施例待测样品锌电解液、锌电解新液、中性上清液由湖南省株洲冶炼厂提供。
实施例1 痕量(较低浓度)钴测定体系的线性回归曲线的制作方法
(1)取Co2+标液(标准溶液)用二次蒸馏水稀释成适宜浓度的Co2+。
(2)分别量取2.5×10-7、5.0×10-7、1.0×10-6、2.0×10-6、4.0×10-6、8.0×10-6和1.6×10-5mol/L的Co2+标液各0.10mL于7个50mL容量瓶中,分别向容量瓶中加入2.6mol/L Zn2+0.10mL,丁二酮肟的乙醇溶液1.4mL,晃动1min,然后依次加入EDTA的氢氧化钠溶液1.0mL,氨-氯化铵缓冲液8.0mL,亚硝酸钠溶液4.0mL于11个50mL容量瓶中,静止5min,分别把溶液倒入电解杯中测定产生的Co2+络合物吸附极谱波,获得测得各浓度Co2+对应的二阶导数波峰电流,绘制线性回归曲线如图1所示(横坐标表示加入容量瓶的Co2+的质量/μg,纵坐标表示二阶导数波峰电流/nA)。
实施例2 微量(较高浓度)钴测定体系的线性回归曲线的制作方法
(1)取Co2+标液(标准溶液)用二次蒸馏水稀释成适宜浓度的Co2+。
(2)分别量取1.0×10-5、2.0×10-5、4.0×10-5、8.0×10-5、4.0×10-6、1.6×10-4、3.0×10-4、3.2×10-4、4.0×10-4、5.0×10-4和6.0×10-4mol/L Co2+标液各0.10mL于11个50mL容量瓶中,分别向容量瓶中加入2.6mol/L Zn2+0.10mL,丁二酮肟的乙醇溶液1.4mL,晃动1min,然后依次加入EDTA的氢氧化钠溶液1.0mL,氨-氯化铵缓冲液8.0mL,静止5min,分别把溶液倒入电解杯中测定产生的Co2+络合物吸附极谱波,获得各浓度Co2+对应的二阶导数波峰电流,绘制线性回归曲线如图2(横坐标表示加入容量瓶的Co2+的质量/mg,纵坐标表示二阶导数波峰电流/nA)所示。
实施例3 采用体系一测定锌电解新液中Co2+
(1)量取0.10mL的锌电解新液于50mL容量瓶中,加入丁二酮肟的乙醇溶液1.4mL,晃动1min,然后依次加入EDTA的氢氧化钠溶液1.0mL,氨-氯化铵缓冲液8.0mL,亚硝酸钠溶液4.0mL,静止5min,把溶液倒入电解杯中测定产生的Co2+络合物吸附极谱波,获得二阶导数波峰电流I1,检测结果如图3所示(横坐标表示扫描电压/mV,纵坐标表示二阶导数波峰电流/nA)。
(2)量取3.0×10-6mol/L Co2+标液0.10mL、0.10mL锌电解新液于50mL容量瓶中,加入丁二酮肟的乙醇溶液1.4mL,晃动1min,然后依次加入EDTA的氢氧化钠溶液1.0mL,氨-氯化铵缓冲液8.0mL,亚硝酸钠4.0mL,静止5min,把溶液倒入电解杯中测定产生的Co2+络合物吸附极谱波,获得二阶导数波峰电流I2。
(3)锌电解新液中Co2+的浓度为3.0×10-6I1/(I2–I1)(mol/L)。具体检测结果数据见表1。
实施例4 采用体系二测定中性上清液中Co2+
(1)量取0.10mL的中性上清液于50mL容量瓶中,加入丁二酮肟的乙醇溶液1.4mL,晃动1min,然后依次加入EDTA的氢氧化钠溶液1.0mL,氨-氯化铵缓冲液8.0mL,静止5min,把溶液倒入电解杯中测定产生的Co2+络合物吸附极谱波,获得二阶导数波峰电流I1,检测结果如图4所示(横坐标表示扫描电压/mV,纵坐标表示二阶导数波峰电流/nA)。
(2)量取6.0×10-4mol/L Co2+标液0.10mL、0.10mL中性上清液于50mL容量瓶中,加入丁二酮肟的乙醇溶液1.4mL,晃动1min,然后依次加入EDTA的氢氧化钠溶液1.0mL,氨-氯化铵缓冲液8.0mL,静止5min,把溶液倒入电解杯中测定产生的Co2+络合物吸附极谱波,获得二阶导数波峰电流I2。
(3)中性上清液中Co2+的浓度为6.0×10-4I1/(I2–I1)(mol/L)。具体检测结果数据见表2。
实施例5 采用体系一测定中性上清液中Co2+
(1)量取1.0mL的中性上清液于1000mL容量瓶中,定容,此为待测溶液。配置3.0×10-3mol/L EDTA的氢氧化钠溶液备用,其余试剂浓度同上。
(2)量取1.0mL步骤(1)待测溶液于50mL容量瓶中,加入丁二酮肟的乙醇溶液1.4mL,晃动1min,然后依次加入步骤(1)所配EDTA的氢氧化钠溶液1.0mL,氨-氯化铵缓冲液8.0mL,亚硝酸钠4.0mL,静止5min,把溶液倒入电解杯中测定产生的Co2+络合物吸附极谱波,获得二阶导数波峰电流I1。检测结果如图5所示(横坐标表示扫描电压/mV,纵坐标表示二阶导数波峰电流/nA)。
(3)量取8.0×10-6mol/L Co2+标液0.10mL,1.0mL的步骤(1)待测溶液于50mL容量瓶中,加入丁二酮肟的乙醇溶液1.4mL,晃动1min,然后依次加入步骤(1)所配EDTA的氢氧化钠溶液1.0mL,氨-氯化铵缓冲液8.0mL,亚硝酸钠4.0mL,静止5min,把溶液倒入电解杯中测定产生的Co2+络合物吸附极谱波,获得二阶导数波峰电流I2。
(4)中性上清液中Co2+的浓度为8.0×10-6I1/(I2-I1)(mol/L)。具体检测结果数据见表3。
实验例1 本发明方法精密度及其与亚硝基R盐分光光度法(NRS)的比较
1.采用体系一测定锌电解新液中Co2+浓度的精密度以及与NRS法测定结果的比较
按实施例3方法进行测定,重复5次。本法五次平行测定值如表1所示,相对标准偏差(RSD)1.1%;结果平均值5.80×10-6mol/L。取相同的锌电解新液样品按照经典方法-亚硝基R盐分光光度法(NRS)【标准号:GB/T 223.22-1994】法进行测定,结果见下表1。本发明方法相对于测定锌电解液中钴的(NRS)测定结果5.74×10-6mol/L偏高1.05%。
表1采用体系一测定锌电解新液中Co2+的结果及其精密度,以及与NRS法测定结果的比较(平行测定5次)
2.采用体系二测定中性上清液中Co2+浓度的精密度以及与NRS法测定结果的比较
按实施例4方法进行测定,重复5次。本法五次平行测定值如表2所示,RSD为0.85%;结果平均值6.50×10-4mol/L。取相同的中性上清液样品按照NRS法进行测定,结果见下表2。本发明方法相对于NRS法测定结果6.57×10-4mol/L偏低1.07%。
表2采用体系二测定中性上清液中Co2+的结果及其精密度,以及与NRS法测定结果的比较(平行测定5次)
3.采用体系一测定中性上清液中Co2+浓度的精密度以及与NRS法测定结果的比较
按实施例5方法进行测定,重复5次。本法五次平行测定值如表3所示,RSD为0.81%;结果平均值6.49×10-4mol/L。取相同的中性上清液样品按照NRS法进行测定,结果见下表3。本发明方法相对于NRS法测定结果6.51×10-4mol/L偏低0.31%。
表3采用体系一测定中性上清液中Co2+的结果及其精密度,以及与NRS法测定结果的比较(平行测定5次)
由表1、表2和表3中结果可知,该方法两个体系对测定不同电解锌液中钴的相对标准偏差≤1.1%,满足方法精密度要求;该方法两个体系测定相同样品中钴的相对偏差为0.15%,相对于测定锌电解液中钴的经典方法-亚硝基R盐分光光度法测定结果偏差的绝对值≤1.07%,满足方法准确度要求。
虽然,上文中已经用一般性说明及具体实施方案对本发明作了详尽的描述,但在本发明基础上,可以对之作一些修改或改进,这对本领域技术人员而言是显而易见的。因此,在不偏离本发明精神的基础上所做的这些修改或改进,均属于本发明要求保护的范围。
- 还没有人留言评论。精彩留言会获得点赞!