一种植物样品中多种内源性植物激素的前处理方法及同时定量检测方法与流程
本发明涉及一种植物样品中多种内源性植物激素的前处理方法及同时定量检测方法,属于分析化学领域。
背景技术:
植物激素在植物生长、发育的各个阶段发挥着及极其重要的作用。同时、准确的定量植物组织中多种植物激素,有助于了解植物激素的生理功能和调控网络。但是,由于植物提取液中基质复杂、含量极低且化学性质各异,使得同时测定植物样品中多种植物激素具有较大困难。
目前已报道的植物中多种植物激素的前处理方法包括液液萃取、固相萃取、固相微萃取、磁固相萃取和液相色谱纯化方法等,这些方法前处理过程多数会带来溶剂消耗大和成本高等问题。此外,由于化学性质差异较大,上述前处理方法会不可避免地造成回收率低,检测灵敏度差,不利于实际样品中的检测。
现有专利ZL 2012 1 0486507.3和ZL 2013 1 0131422.8,是基于油菜素甾醇(BRs)的分析检测。由于BRs类激素化学结构性质相似,且都含有顺式羟基,针对这一类物质的性质,有了上述专利所述的前处理方案。但是对于化学性质不同于油菜素甾醇(BRs)的其他类植物激素,用上述方案则无法实现。
技术实现要素:
本发明所要解决的技术问题是针对上述现有技术存在的不足而提供一种植物样品中多种内源性植物激素的前处理方法及同时定量检测方法,可同时实现低含量的多种内源性植物激素的富集纯化,并实现同时定量检测,方法简单快速,准确性高。
本发明为解决上述提出的问题所采用的技术方案为:
一种植物样品中多种内源性植物激素的前处理方法,主要步骤如下:
1)在植物样品中定量加入多种内源性植物激素的同位素内标,然后以溶剂提取得到样品溶液;
2)以石墨化炭黑材料为净化介质对步骤1)所得样品溶液进行除杂,进而得到植物样品中多种内源性植物激素的待测溶液,从而实现植物样品中多种内源性植物激素的样品前处理。
按上述方案,所述植物样品为幼苗期的水稻,分蘖期的水稻,灌浆期的叶片和花穗,成熟期的叶片和花穗,以及水稻的根,拟南芥的根、茎、叶、花和角果等等植物样品。
按上述方案,所述植物样品为粉末状,如可以预先采用液氮冷冻后研磨至粉末状。
按上述方案,所述内源性植物激素的种类主要包括生长素类、细胞分裂素类、赤霉素类、茉莉酸类、水杨酸类以及脱落酸类的内源性植物激素等等。
按上述方案,所述内源性植物激素主要包括吲哚-3-乙酸(IAA),吲哚-3-丁酸(IBA),脱落酸(ABA),水杨酸(SA),反式玉米素(tZ),反式玉米素核苷(tZR),顺式玉米素(cZ),顺式玉米素核苷(cZR),异戊烯基腺嘌呤(iP),异戊烯基腺嘌呤核苷(iPR),双氢玉米素(DZ),双氢玉米素核苷(DZR),反式玉米素-7-糖苷(tZ7G),反式玉米素-9-糖苷(tZ9G),顺式玉米素-9-糖苷(cZ9G),顺式玉米素-O-糖苷(cZOG),双氢玉米素-7-糖苷(DZ7G),双氢玉米素-9-糖苷(DZ9G),双氢玉米素-O-糖苷(DZOG),异戊烯基腺嘌呤-7-糖苷(iP7G),异戊烯基腺嘌呤-9-糖苷(iP9G),2-苄硫基异戊烯基腺嘌呤(2BSiP),2-氯反式玉米素(2CltZ),2-甲硫基反式玉米素(2MeStZ),2-甲硫基顺式玉米素(2MeScZ),2-甲硫基反式玉米素核苷(2MeStZR),2-甲硫基顺式玉米素核苷(2MeScZR),2-甲硫基异戊烯基腺嘌呤(2MeSiP),2-甲硫基异戊烯基腺嘌呤核苷(2MeSiPR),赤霉素1(GA1),赤霉素3(GA3),赤霉素4(GA4),赤霉素5(GA5),赤霉素6(GA6),赤霉素7(GA7),赤霉素8(GA8),赤霉素9(GA9),赤霉素12(GA12),赤霉素13(GA13),赤霉素15(GA15),赤霉素19(GA19),赤霉素20(GA20),赤霉素23(GA23),赤霉素24(GA24),赤霉素29(GA29),赤霉素34(GA34),赤霉素44(GA44),赤霉素51(GA51),赤霉素53(GA53),茉莉酸(JA),茉莉酸-异亮氨酸(JA-leu),茉莉酸-苯丙氨酸(JA-phe),12-羟基茉莉酸(12OHJA),12-氧代植二烯酸(OPDA),脱落酸(ABA)和水杨酸(SA)等等中的一种或多种。
按上述方案,根据需要加入相应的同位素内标,主要为[2H2]IAA,[2H6]ABA,二氢茉莉酸(2H-JA),[2H4]SA,[2H2]GA1,[2H2]GA4,[2H2]GA5,[2H2]GA6,[2H2]GA7,[2H2]GA8,[2H2]GA9,[2H2]GA12,[2H2]GA15,[2H2]GA20,[2H2]GA24,[2H2]GA34,[2H2]GA44,[2H2]GA51,[2H2]GA53,[2H5]tZ,[15N5]cZ,[2H5]tZ7G,[2H5]tZ9G,[2H3]DZ,[2H5]DZ9G,[2H7]DZOG,[2H6]iP,[2H6]iP9G,[2H5]tZR,[2H3]DZR,[2H6]iPR等等。待测多种内源性植物激素与之相对应的内标如表1所示。
按上述方案,所述溶剂为甲醇、乙腈、异丙醇、氯仿、二氯甲烷、乙酸乙酯、正己烷、乙醚、丙酮或甲酸等中的一种或几种按任意比例的混合物。溶剂的作用是:在有效地提取出分析物的前提下,尽可能少地提取杂质等干扰物。
按上述方案,所述溶剂提取的温度为-25~-15℃,时间范围为6~12h。
按上述方案,所述加入石墨化炭黑材料后除杂的温度为室温,时间为3min或者大于3min。除杂过程中可以采用涡旋或者振荡等方式以保证除杂均匀充分。
按上述方案,步骤2)中优选地步骤为:首先,除去步骤1)所得样品溶液中的溶剂,复溶后加入石墨化炭黑材料进行除杂,收集除杂后所得上清液,即为植物样品中多种内源性植物激素的待测溶液。
按上述方案,步骤2)中更优选地步骤为:首先,除去步骤1)所得样品溶液中的溶剂,得到样品渣;然后所得样品渣加入溶剂B复溶,得到复溶后的样品溶液;接着,将所得复溶后的样品溶液中加入石墨化炭黑材料进行除杂,收集除杂后所得上清液;最后,将除杂后所得上清液中的溶液B除去,加入溶剂C复溶,得到植物样品中多种内源性植物激素的待测溶液。其中,溶剂B为可以溶解样品渣,且不吸附或者少吸附目标分析物多种内源性植物激素的溶剂,例如甲醇及其与水的混合溶液、乙腈及其与水的混合溶液等;所述溶剂C能与后续的分离条件相适应,例如甲醇及其与水的混合溶液、乙腈及其与水的混合溶液等。
按上述方案,所述步骤2)中的石墨化炭黑材料为含有π-π共轭基团的炭基颗粒及包含该种炭基颗粒的凝胶、磁性材料、无定型材料、填充柱或整体柱等中的一种或几种。所述的除杂是指除去样品溶液中含有的大量色素、脂质和强疏水性杂质,故采用由碳材料为基底,构建的硅胶颗粒、填充柱或整体柱等均可以达到除杂的目的。
本发明在上述前处理方法的基础上还提供一种植物样品中多种内源性植物激素的同时定量检测方法,将前处理方法所得植物样品中多种内源性植物激素的待测溶液通过分析仪器采集响应信号或者数据,进而采用内标法同时定量测定植物样品中的多种内源性植物激素。
按上述方案,所述分析仪器可以采用液相色谱-光学检测器、液相色谱-质谱仪联用、毛细管电泳-光学检测器联用、毛细管电泳-质谱仪联用等。优选地,所述分析仪器采用高效液相色谱-电喷雾-三重四级杆串联质谱。
具体地,一种植物样品中多种内源性植物激素的同时定量检测方法,主要步骤如下:
1)配置不同浓度梯度的标准样品溶液,并定量加入多种内源性植物激素的同位素内标,然后对所配置好的标准溶液进行超高效液相色谱-串联质谱(UPLC-MS/MS)分析得到色谱图,然后将每个物质(即每种内源性植物激素)的色谱图峰面积积分,除以与之相应的内标的峰面积,对相应物质(即每种内源性植物激素)的浓度作线性回归,制得标准曲线;
2)在植物样品中定量加入多种内源性植物激素的同位素内标,然后以溶剂提取得到样品溶液;
3)以石墨化炭黑材料为净化介质对步骤2)所得样品溶液进行除杂,进而得到植物样品中多种内源性植物激素的待测溶液;
4)将所得植物样品中多种内源性植物激素的待测溶液通过超高效液相色谱-串联质谱(UPLC-MS/MS)分析采集色谱图;进而将色谱图中每个物质(即每种内源性植物激素)的峰面积积分,除以与之相应的内标的峰面积,采用步骤1)所得标准曲线同时定量测定植物样品中的多种内源性植物激素。
与现有技术相比,本发明的有益效果是:
1、本发明方法简单、快速、省溶剂、易操作,可实现低含量的内源性植物激素的富集纯化,并实现植物样品中多种内源性植物激素的同时定量检测,方法简单快速,准确性高。
2、与背景技术中所提到的现有技术相比,本发明可以同时实现高达54种及以上的性质各异的植物激素的分析检测,而不是仅适用于一类植物激素(油菜素甾醇)检测。
3、与背景技术中所提到的现有技术相比,本发明中石墨化炭黑能够实现植物提取液的净化,而不吸附目标分析物---性质各异的植物激素,取得了意料不到的技术效果;而背景技术中所提到专利ZL 2012 1 0486507.3中,石墨化炭黑仅作为前处理的一部分,方案复杂,且吸附较多非油菜素甾醇类植物激素。
4、本发明能采用一步前处理的方案,减少了目标分析物在前处理中的损失;同时,采用分离更加高效的超高效液相系统,可以分离众多分子量相同的化合物,以此实现了54种植物激素的检测。
附图说明
图1为本发明方法可以实施分析的54种植物激素的多反应监控色谱图。其中,(A),peak 1-54;(B),peak1-7;(C),peak22-27;(D),peak38-42;1,tZ7G;2,tZ;3,DZ;4,cZOG;5,DZ7G;6,DZOG;7,cZ;8,DZ9G;9,tZ9G;10,cZ9G;11,iP7G;12,iP;13,DZR;14,tZR;15,GA8;16,cZR;17,GA29;18,iP9G;19,12OHJA;20,GA23;21,SA;22,2CltZ;23,GA3;24,iPR;25,2MeStZ;26,2MeScZ;27,GA1;28,IAA;29,GA6;30,2MeStZR;31,2MeStZR;32,ABA;33,GA13;34,GA5;35,GA19;36,GA20;37,GA44;38,JA;39,IBA;40,GA34;41,2MeSiP;42,GA51;43,GA53;44,2MeSiPR;45,GA7;46,GA4;47,GA24;48,JA-leu;49,JA-phe;50,GA15;51,GA9;52,2BSiP;53,GA12;54,OPDA。
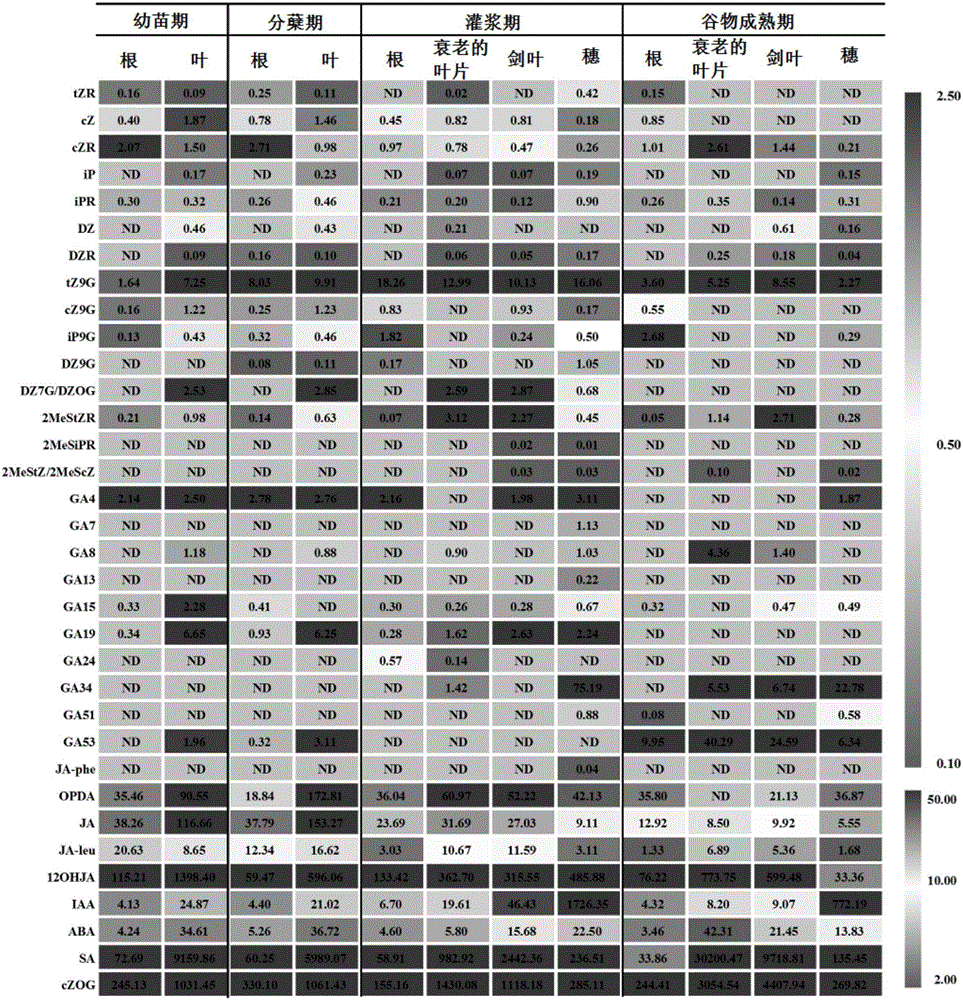
图2为本发明法检测到的不同植物组织中内源性植物激素的含量。其中(A)为植物激素在水稻样品中的分布热图;(B)细胞分类素和赤霉素在水稻中的总含量以及不同类型的细胞分裂素所占比例。
具体实施方式
为了更好地理解本发明,下面结合实施例进一步对本发明的所提供技术方案中的关键技术作进一步的说明,但本发明不仅仅局限于下面的实施例。
实施例1
一种植物样品中多种内源性植物激素的前处理方法,主要步骤如下:
(1)准确称取50mg植物组织(分别平行取幼苗期的水稻,分蘖期的水稻,灌浆期的叶片和花穗,成熟期的叶片和花穗,以及水稻的根这些不同部位的植物组织作为前处理以及后续定量检测的植物样品),转移至1.5mL离心管中,定量加入多种内源性植物激素的同位素内标[2H2]IAA(1.0ng),[2H6]ABA(1.0ng),2H-JA(1.0ng)[2H4]SA(50ng),[2H2]GA1(0.5ng),[2H2]GA4(0.5ng),[2H2]GA5(0.5ng),[2H2]GA6(0.5ng),[2H2]GA7(0.5ng),[2H2]GA8(0.5ng),[2H2]GA9(0.5ng),[2H2]GA12(0.5ng),[2H2]GA15(0.5ng),[2H2]GA20(0.5ng),[2H2]GA24(0.5ng),[2H2]GA34(0.5ng),[2H2]GA44(0.5ng),[2H2]GA51(0.5ng),[2H2]GA53(0.5ng),[2H5]tZ(0.1ng),[2H5]cZ(0.1ng),[2H5]tZ7G(0.1ng),[2H5]tZ9G(0.1ng),[2H3]DZ(0.1ng),[2H5]DZ9G(0.1ng),[2H7]DZOG(0.1ng),[2H6]iP(0.1ng),[2H6]iP9G(0.1ng),[2H5]tZR(0.1ng),[2H3]DZR(0.1ng),[2H6]iPR(0.1ng),放置片刻后,加入0.5mL乙腈混合均匀,放入-20℃的冰箱内提取12小时,离心取上清液,即为样品溶液;
(2)除去步骤(1)所得样品溶液中的溶剂,用80%(v/v)乙腈水溶液复溶,将所得复溶后的样品溶液加入到含有10mg石墨化炭黑的1.5mL的离心管中,涡旋3分钟进行除杂,离心取上清液并吹干以除去溶剂,再用100微升5%的乙腈水溶液复溶,即得到植物样品中多种内源性植物激素的待测溶液,从而实现植物样品中多种内源性植物激素的样品前处理。
一种植物样品中多种内源性植物激素的同时定量检测方法,主要步骤如下:
1)配置不同浓度梯度的标准样品溶液(浓度范围如表1所示),并加入内标[2H2]IAA(1.0ng),[2H6]ABA(1.0ng),2H-JA(1.0ng)[2H4]SA(50ng),[2H2]GA1(0.5ng),[2H2]GA4(0.5ng),[2H2]GA5(0.5ng),[2H2]GA6(0.5ng),[2H2]GA7(0.5ng),[2H2]GA8(0.5ng),[2H2]GA9(0.5ng),[2H2]GA12(0.5ng),[2H2]GA15(0.5ng),[2H2]GA20(0.5ng),[2H2]GA24(0.5ng),[2H2]GA34(0.5ng),[2H2]GA44(0.5ng),[2H2]GA51(0.5ng),[2H2]GA53(0.5ng),[2H5]tZ(0.1ng),[2H5]cZ(0.1ng),[2H5]tZ7G(0.1ng),[2H5]tZ9G(0.1ng),[2H3]DZ(0.1ng),[2H5]DZ9G(0.1ng),[2H7]DZOG(0.1ng),[2H6]iP(0.1ng),[2H6]iP9G(0.1ng),[2H5]tZR(0.1ng),[2H3]DZR(0.1ng),[2H6]iPR(0.1ng);
2)对步骤1)配置好的标准溶液进行超高效液相色谱-串联质谱(UPLC-MS/MS)分析,得如图1所示的色谱图,然后将每个物质的色谱图峰面积积分,除以与之相应的内标的峰面积,对相应物质的浓度作线性回归,制得标准曲线(如表1所示);
3)将本实施例前处理方法所得不同水稻组织部位的植物激素的待测溶液通过超高效液相色谱-串联质谱(UPLC-MS/MS)分析采集色谱图;进而将色谱图中每个物质的峰面积积分,除以与之相应的内标的峰面积,采用步骤2)所得标准曲线同时定量测定植物样品中的多种内源性植物激素,测得不同水稻组织部位的植物激素含量如图1所示。
表1 分析物的工作曲线
为了验证方法的准确度,本发明向水稻样品中加入不同浓度的细胞分裂素标品(低、中、高,详见表2),之后按照所述的方法进行处理与测定,得出不同加标浓度的回收率在80.3%-120.4%之间,表明本发明所提供方法的准确性。为了考察方法的精密度,在一天内对不同加标浓度的样品进行5次重复处理与测定,计算各自的回收率的相对标准偏差(RSD),即为日内精密度;连续三天对不同加标浓度的样品进行处理与测定,计算各自的回收率的相对标准偏差(RSD),即为日间精密度。结果如表2所示,RSD小于11.8%,结果表明本发明所述方法稳定可靠。
表2 方法的稳定度和准确度
综上所述,本发明能采用一步前处理的方案,可实现低含量的内源性植物激素的富集纯化,简单、快速、省溶剂、易操作,并后续可实现高达54种及以上的性质各异的植物激素的同时定量检测,方法简单快速,准确性高,完全跳出了现有技术中一种前处理及其定量检测方法仅能针对一类植物激素的局限。
以上所述仅是本发明的优选实施方式,应当指出,对于本领域的普通技术人员来说,在不脱离本发明创造构思的前提下,还可以做出若干改进和变换,这些都属于本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!