一种药物中阳离子表面活性剂的分离检测方法与流程
本发明属于分析测定领域,涉及一种分离检测方法,尤其涉及一种药物中阳离子表面活性剂的分离检测方法。
背景技术:
阳离子表面活性剂在水溶液中电离时生成的表面活性离子带正电荷,其疏水基与阴离子表面活性剂相似。阳离子表面活性剂的亲水基离子中含有氮原子,根据氮原子在分子中的位置不同分为胺盐、季铵盐和杂环型三类。
季铵盐阳离子表面活性剂水溶性好,既耐酸又耐碱且大多数具有杀菌作用。用季铵盐阳离子表面活性剂可与许多物质亲和、吸附形成氢键,具有除浊、脱色、吸附和粘合等功能,适用于发酵制药等行业中的纯化工艺,应用非常广泛。
玻璃酸钠为一种原料药,其工艺生产中使用十六烷基三甲基氯化铵(CTAC)这种季铵盐类阳离子表面活性剂作为络合剂,鉴于产品的使用安全性考虑,在产品中残留量应严格控制,因此如何实现对药物中铵盐阳离子表面活性剂等表面活性剂的分离和检测是十分重要的。
对于大多数铵盐类阳离子表面活性剂因其具有不易挥发、极性强等特点,通常采用液相色谱进行定量分析,例如采用连接DAD检测器(二极管阵列检测器)的高效液相色谱方法进行定量分析,但是如烷基铵盐类阳离子表面活性剂没有生色团,因此不能直接用常规的连接DAD检测器或荧光检测器的高效液相色谱仪器进行分析。
研究表明,采用连接电导检测器的高效液相色谱分析方法可对没有生色团的烷基铵盐类阳离子表面活性剂进行定量检测。如CN 102226796A公开了一种测定两性表面活性剂中微量N,N-二甲基丙二胺的分析方法,该方法采用离子色谱法进行分析,色谱检测条件如下:色谱柱:阳离子色谱柱;流动相:由体积百分含量为70%~100%的无机酸或有机酸酸水溶液和体积百分含量为0%~30%的有机溶剂组成,所述无机酸或有机酸水溶液的摩尔浓度为1mmol/L~10mmol/L;检测器:采用电导检测器;流动相流速:0.5mL/min~1.5mL/min。
另外通过加入有机试剂如二溴羧基苯基重氮氨基偶氮苯,在碱性介质中与烷基铵盐类阳离子表面活性剂发生缔合显色反应,然后通过分光光度法进行分析也是一种检测烷基铵盐类阳离子表面活性剂的方法。
然而,上述方法在存在干扰物质时具有定性定量效果差的缺点。
采用大气压电喷雾质朴法(ESI/MS)连接高效液相色谱进行分析,根据质谱提供的分子量信息对其结构及组成进行定性,利用高效液相色谱进行定量分析,可准确定性和定量化两种或多种混合表面活性剂,但此法所用检测仪器价格昂贵,成本较高。
并且,由于药物中组成比较复杂,在对表面活性剂进行分离测定时由于其他组分的干扰,很难做到对表面活性剂精确的分离和测定。
因此,如何寻求一种可以有效测定药物中阳离子表面活性剂,尤其是烷基铵盐类阳离子表面活性剂,且成本小的方法是非常有必要的。
技术实现要素:
针对现有阳离子表面活性剂分析测定方法中存在的定性定量效果差,且仪器价格昂贵,成本高,以及目前针对药物中表面活性剂等物质分离和测定困难等问题,本发明提供了一种药物中阳离子表面活性剂的分离检测方法。本发明所述分离检测方法通过配置合理浓度的样品溶液,调节气相色谱仪的工作参数,可以有效避免药物中其他成分的干扰,精密度高,重复性好且成本低。
为达此目的,本发明采用以下技术方案:
本发明提供了一种药物中阳离子表面活性剂的分离检测方法,所述方法包括以下步骤:
(1)配制药物样品溶液;
(2)将步骤(1)配制的药物样品溶液注入气相色谱仪,采用如下条件对药物样品溶液中的阳离子表面活性剂进行分离测定;
所述色谱条件为:
以氮气为载气,控制载气流量为2mL/min~8mL/min,例如2mL/min、3mL/min、4mL/min、5mL/min、6mL/min、7mL/min或8mL/min等,但并不仅限于所列举的数值,该数值范围内其他未列举的数值同样适用。
气相色谱仪进样口温度为250℃~300℃,例如250℃、260℃、270℃、280℃、290℃或300℃等,但并不仅限于所列举的数值,该数值范围内其他未列举的数值同样适用。
气相色谱仪柱温为:在起始温度90℃~150℃下维持2min~5min,再以10℃/min~15℃/min的速率升温至240℃~260℃,维持4min~7min。其中,起始温度可为90℃、100℃、110℃、120℃、130℃、140℃或150℃等,但并不仅限于所列举的数值,该数值范围内其他未列举的数值同样适用;起始温度维持时间可为2min、3min、4min或5min等,但并不仅限于所列举的数值,该数值范围内其他未列举的数值同样适用;升温速率可为10℃/min、11℃/min、12℃/min、13℃/min、14℃/min或15℃/min等,但并不仅限于所列举的数值,该数值范围内其他未列举的数值同样适用;升温后的温度可为240℃、245℃、250℃、255℃或260℃等,但并不仅限于所列举的数值,该数值范围内其他未列举的数值同样适用;升温后维持时间可为4min、5min、6min或7min等,但并不仅限于所列举的数值,该数值范围内其他未列举的数值同样适用。
FTD检测器温度为250℃~320℃,例如250℃、260℃、270℃、280℃、290℃、300℃、310℃或320℃等,但并不仅限于所列举的数值,该数值范围内其他未列举的数值同样适用。
本发明所述气相色谱检测分离方法中的色谱条件对于检测结果起到决定性作用,其中载气流量、气相色谱仪进样口温度以及柱温均需控制在一定的条件下,才可保证测试的准确性。
本发明中,所述载气流量需控制在2mL/min~8mL/min内,若载气流量过低,会使目标峰保留时间较小,分离不完全,降低测试的准确性;若载气流量过高,会使目标峰保留时间较大,检测时间过长。
所述进样口温度需控制在250℃~300℃内,若进样口温度过低,会使检测信号强度小,测试的准确性;若进样口温度过高,会使毛细管柱端损坏。
所述气相色谱仪柱温的起始温度需控制在90℃~150℃内,过高会使目标峰保留时间较小,分离不完全,降低测试的准确性;过低会使目标峰保留时间较大,检测时间过长。同样的,其检测温度也需控制在250℃~320℃内,过高会使毛细管柱端损坏;过低会使检测信号强度小。
以下作为本发明优选的技术方案,但不作为本发明提供的技术方案的限制,通过以下技术方案,可以更好的达到和实现本发明的技术目的和有益效果。
作为本发明优选的技术方案,所述药物为发酵方法制得的原料药及其制剂和/或合成方法制得的原料药及其制剂,优选为发酵法制得的原料药及其制剂,进一步优选为玻璃酸钠。
优选地,所述制剂为注射剂。
作为本发明优选的技术方案,所述阳离子表面活性剂为烷基季铵盐类阳离子表面活性剂。
优选地,所述烷基季铵盐类阳离子表面活性剂为十二烷基三甲基氯化铵、十六烷基三甲基氯化铵或十八烷基三甲基氯化铵中任意一种或至少两种的组合,所述组合典型但非限制性实例有:十二烷基三甲基氯化铵和十六烷基三甲基氯化铵的组合,十六烷基三甲基氯化铵和十八烷基三甲基氯化铵的组合,十二烷基三甲基氯化铵、十六烷基三甲基氯化铵和十八烷基三甲基氯化铵的组合等,优选为十六烷基三甲基氯化铵。
作为本发明优选的技术方案,步骤(1)所述配制药物样品溶液为:将药物与稀释剂混合制成药物样品浓度为3mg/mL~10mg/mL的药物样品溶液,例如3mg/mL、4mg/mL、5mg/mL、6mg/mL、7mg/mL、8mg/mL、9mg/mL或10mg/mL等,但并不仅限于所列举的数值,该数值范围内其他未列举的数值同样适用,优选为药物样品浓度为5mg/mL的药物样品溶液。
本发明中,药物样品浓度也需控制在一定范围内,若溶浓度过高,会使目标峰响应值过大而使其与相邻杂质分离不完全;若浓度过低会使目标峰响应值过小影响检测准确度。
优选地,所述稀释剂为乙醇和水的混合物。
优选地,所述乙醇和水的混合物为体积比为(5~1):(1~5)的混合物,例如5:(1~5)、4:(1~5)、3:(1~5)、2:(1~5)或1:(1~5)等,又如(5~1):1、(5~1):2、(5~1):3、(5~1):4或(5~1):5等,更进一步的可5:1、4:2、3:4、2:3、2:1或1:5等,但并不仅限于所列举的数值,该数值范围内其他未列举的数值同样适用,优选为体积比为1:1的混合物。
作为本发明优选的技术方案,步骤(2)所述分离测定过程中气相色谱仪中毛细管柱中的固定液为聚硅氧烷,优选为甲基聚硅氧烷。
作为本发明优选的技术方案,步骤(2)所述分离测定过程中气相色谱仪中所通氢气的流量为20mL/min~50mL/min,例如20mL/min、30mL/min、40mL/min或50mL/min等,但并不仅限于所列举的数值,该数值范围内其他未列举的数值同样适用;所通空气的流量为200mL/min~500mL/min,例如200mL/min、300mL/min、400mL/min或500mL/min等,但并不仅限于所列举的数值,该数值范围内其他未列举的数值同样适用。
作为本发明优选的技术方案,步骤(2)所述分离测定过程中色谱条件为:以氮气为载气;控制载气流量为4mL/min~6mL/min;气相色谱仪进样口温度为275℃~285℃;气相色谱仪柱温为:在起始温度115℃~125℃下维持2min~3min,再以10℃/min~12℃/min的速率升温至245℃~255℃,维持4min~5min。
优选地,步骤(2)所述分离测定过程中色谱条件为:以氮气为载气;控制载气流量为5mL/min;气相色谱仪进样口温度为280℃;气相色谱仪柱温为:在起始温度120℃下维持2min,再以10℃/min的速率升温至250℃,维持4min。
本发明中分离测定过程中色谱条件对于检测结果起到决定性作用,其又以上述测定条件下测得的结果更优。
作为本发明优选的技术方案,所述药物为玻璃酸钠时,其分离检测方法包括以下步骤:
(1’)配制玻璃酸钠样品溶液;
(2’)将步骤(1’)配制的玻璃酸钠样品溶液注入气相色谱仪,采用如下条件对玻璃酸钠样品溶液中的十六烷基三甲基氯化铵进行分离测定;
所述色谱条件为:
毛细管柱中的固定液为甲基聚硅氧烷;
以氮气为载气,控制载气流量为5mL/min;
气相色谱仪进样口温度为280℃;
气相色谱仪柱温为:在起始温度120℃下维持2min,再以10℃/min的速率升温至250℃,维持4min;
FID检测器的温度为300℃,其所通氢气的流量为30mL/min,所通空气的流量为300mL/min。
本发明对于药物玻璃酸钠中十六烷基三甲基氯化铵的分离测定,以上述色谱条件进行分离测定的结果最优。
作为本发明优选的技术方案,步骤(1’)所述配制药物样品溶液为:将药物与稀释剂混合制成药物样品浓度为5mg/mL的药物样品溶液。
作为本发明优选的技术方案,所述稀释剂为乙醇和水以体积比为1:1组成的混合物。
本发明所述分离检测方法中所用气相色谱仪中所用空白溶液为稀释剂溶液,即为乙醇和水的混合物,所述乙醇和水的混合物为体积比为(5~1):(1~5)的混合物,优选为体积比为1:1的混合物。
气相色谱仪中所用对照样品溶液为阳离子表面活性剂溶于稀释剂制得的溶液,其浓度为0.001mg/mL~0.01mg/mL,优选为0.005mg/mL。
与现有技术相比,本发明具有以下有益效果:
本发明所述分离测试方法通过合理浓度的样品溶液,优化气相色谱仪的工作参数,可有效避免药物中其他组成成分对于阳离子表面活性剂分离测试的干扰,进而可以将阳离子表面活性剂有效的从药物中分离出来,并准确算出药物中阳离子表面活性剂的含量,其回收率可达100%,精密度高,重复性好。且所用气相色谱仪与其他测试仪器相比,价格低廉,成本更低。
附图说明
图1是本发明实施例1中线性测试色谱图;
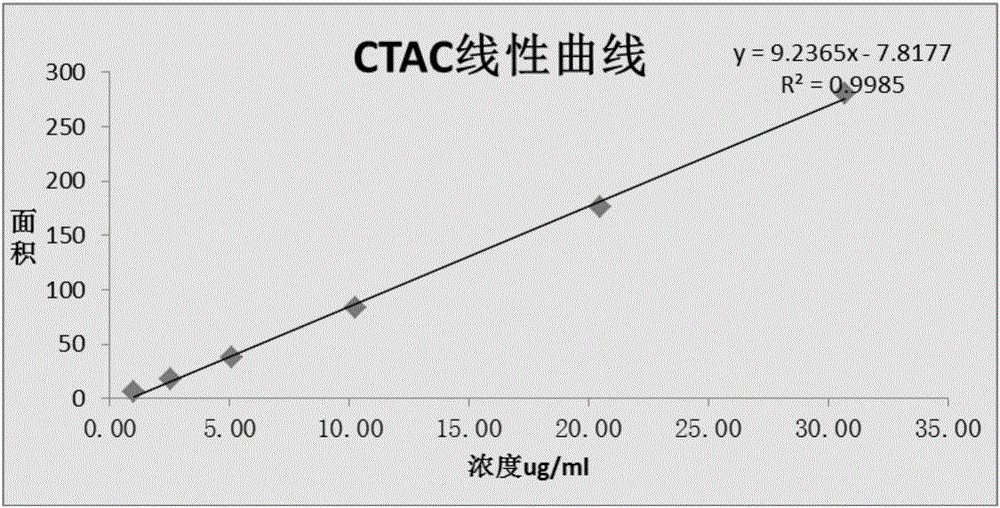
图2是本发明实施例1中线性测试的线性曲线。
具体实施方式
为更好地说明本发明,便于理解本发明的技术方案,下面对本发明进一步详细说明。但下述的实施例仅仅是本发明的简易例子,并不代表或限制本发明的权利保护范围,本发明保护范围以权利要求书为准。
本发明具体实施例部分提供了一种药物中阳离子表面活性剂的分离检测方法,所述方法包括以下步骤:
(1)配制药物样品溶液;
(2)将步骤(1)配制的药物样品溶液注入气相色谱仪,采用如下条件对药物样品溶液中的阳离子表面活性剂进行分离测定;
所述色谱条件为:
以氮气为载气,控制载气流量为2mL/min~8mL/min;
气相色谱仪进样口温度为250℃~300℃;
气相色谱仪柱温为:在起始温度90℃~150℃下维持2min~5min,再以10℃/min~15℃/min的速率升温至240℃~260℃,维持4min~7min。
FTD检测器温度为250℃~320℃。
以下为本发明典型但非限制性实施例:
实施例1:
本实施例提供了一种发酵工艺生产的玻璃酸钠及其制剂中十六烷基三甲基氯化铵的分离测定方法:
本分离测定方法中所用空白溶液为稀释剂,即乙醇与水以体积比50:50配制得到;
所用对照品溶液的配制为:
称取十六烷基三甲基氯化铵25mg置于50mL容量瓶,加稀释剂溶解并稀释至刻度,摇匀,然后移取0.1mL置于另一个10ml容量瓶,用稀释剂稀释至刻度,摇匀,作为对照品溶液。
所用药物样品溶液为:
取样品约50mg,精密称定,置于10mL容量瓶,加稀释剂5mL,振荡5分钟,加稀释剂稀释至刻度,摇匀,静置15min,取溶液离心15min,取上清溶液,作为药物样品溶液。
将步骤配制的药物样品溶液注入气相色谱仪,采用如下条件对药物样品溶液中的阳离子表面活性剂进行分离测定;
毛细管柱中的固定液为甲基聚硅氧烷;
氮气为载气,控制载气流量为5mL/min;
气相色谱仪进样口温度为280℃;
气相色谱仪柱温为:在起始温度120℃下维持2min,再以10℃/min的速率升温至250℃,维持4min。
FID检测器的温度为300℃,其所通氢气的流量为30mL/min,所通空气的流量为300mL/min。
所用毛细管柱为Agilent 30m*0.32mm,0.5um;所述气相色谱仪为Agilent7890A。
分别精密量取1μL上述配制的空白溶液、对照品溶液和药物样品溶液,注入气相色谱仪(仪器型号:Agilent7890A),并记录色谱图。
以外标法测定药物样品溶液中十六烷基三甲基氯化铵的含量,结果详见表1。
表1:药物样品溶液中十六烷基三甲基氯化铵含量检测分析表
注:NA表示不适用。
从表1中可以看出,本方法能使对照品溶液和样品溶液中的阳离子表面活性剂十六烷基三甲基氯化铵达到很好分离,并准确计算出测定药物样品溶液中十六烷基三甲基氯化铵的含量,其回收率可达100%,精密度高,重复性良好。
线性曲线测试:
线性测试溶液的配制:称取十六烷基三甲基氯化铵25mg置于50mL容量瓶,加稀释剂溶解并稀释至刻度,摇匀,然后分别从三个容量瓶中移取0.6mL、0.4mL、0.2mL、0.1mL、0.05mL和0.025mL分别置于10mL容量瓶,用稀释剂稀释至刻度,摇匀,作为6个不同浓度的对照品线性溶液。
分别精密量取1μL上述配制的6个不同浓度的对照品线性溶液,注入气相色谱仪,并采用上述气相色谱条件,并记录色谱图(如图1所示)。
计算测得的峰面积,结果如表2所示。并根据测得的峰面积和实际浓度进行线性分析,并绘制线性曲线(如图2所示)。
表2:线性测试数据结果表
从表2以及图1和图2的结果可以看出,本发明提供的气相色谱方法对样品中阳离子表面活性剂十六烷基三甲基氯化铵的分离测定,在十六烷基三甲基氯化铵浓度为1.02~30.72ug/mL范围内与峰面积具有良好的线性关系。
实施例2:
本实施例提供了一种发酵工艺生产的玻璃酸钠及其制剂中十六烷基三甲基氯化铵的分离测定方法:
本分离测定方法中所用空白溶液为稀释剂,即乙醇与水以体积比50:50配制得到;
所用对照品溶液的配制为:
称取十六烷基三甲基氯化铵25mg置于50mL容量瓶,加稀释剂溶解并稀释至刻度,摇匀,然后移取0.1mL置于另一个10ml容量瓶,用稀释剂稀释至刻度,摇匀,作为对照品溶液。
所用药物样品溶液为:
取样品约50mg,精密称定,置于10mL容量瓶,加稀释剂5mL,振荡5分钟,加稀释剂稀释至刻度,摇匀,静置15min,取溶液离心15min,取上清溶液,作为药物样品溶液。
将步骤配制的药物样品溶液注入气相色谱仪,采用如下条件对药物样品溶液中的阳离子表面活性剂进行分离测定;
毛细管柱中的固定液为聚硅氧烷;
氮气为载气,控制载气流量为4mL/min;
气相色谱仪进样口温度为275℃;
气相色谱仪柱温为:在起始温度115℃下维持3min,再以12℃/min的速率升温至245℃,维持5min;
FID检测器的温度为290℃,其所通氢气的流量为25mL/min,所通空气的流量为250mL/min。
所用毛细管柱为Agilent 30m*0.32mm,0.5um;所述气相色谱仪为Agilent7890A。
分别精密量取1μL上述配制的空白溶液、对照品溶液和药物样品溶液,注入气相色谱仪,并记录色谱图。
以外标法测定药物样品溶液中十六烷基三甲基氯化铵的含量为0.00301%,其回收率可达100.3%,精密度高,重复性良好。
实施例3:
本实施例提供了一种发酵工艺生产的药物及其制剂中十二烷基三甲基氯化铵的分离测定方法:
所用对照品溶液的配制为:
称取十二烷基三甲基氯化铵25mg置于50mL容量瓶,加稀释剂溶解并稀释至刻度,摇匀,然后移取0.1mL置于另一个10ml容量瓶,用稀释剂稀释至刻度,摇匀,作为对照品溶液。
所用药物样品溶液为:
取样品约50mg,精密称定,置于10mL容量瓶,加稀释剂5mL,振荡5分钟,加稀释剂稀释至刻度,摇匀,静置15min,取溶液离心15min,取上清溶液,作为药物样品溶液。
将步骤配制的药物样品溶液注入气相色谱仪,采用如下条件对药物样品溶液中的阳离子表面活性剂进行分离测定;
毛细管柱中的固定液为聚硅氧烷;
氮气为载气,控制载气流量为6mL/min;
气相色谱仪进样口温度为285℃;
气相色谱仪柱温为:在起始温度125℃下维持4min,再以15℃/min的速率升温至255℃,维持5min;
FID检测器的温度为310℃,其所通氢气的流量为35mL/min,所通空气的流量为350mL/min。
所用毛细管柱为Agilent 30m*0.32mm,0.5um;所述气相色谱仪为Agilent 7890A。
分别精密量取1μL上述配制的空白溶液、对照品溶液和药物样品溶液,注入气相色谱仪,并记录色谱图。
以外标法测定药物样品溶液中十二烷基三甲基氯化铵的含量为0.00303%,其回收率可达100.3%,精密度高,重复性良好。
实施例4:
本实施例提供了一种发酵工艺生产的药物及其制剂中十八烷基三甲基氯化铵的分离测定方法,所述方法除了气相色谱仪的条件为:毛细管柱中的固定液为甲基聚硅氧烷;
氮气为载气,控制载气流量为2mL/min;
气相色谱仪进样口温度为250℃;
气相色谱仪柱温为:在起始温度90℃下维持5min,再以10℃/min的速率升温至240℃,维持7min。
FID检测器的温度为250℃,其所通氢气的流量为20mL/min,所通空气的流量为200mL/min。
所用毛细管柱为Agilent 30m*0.32mm,0.5um;所述气相色谱仪为Agilent 7890A。
其他过程均与实施例1中相同,以外标法测定药物样品溶液中十八烷基三甲基氯化铵的含量为0.00302%,其回收率可达100.7%,精密度高,重复性良好。
实施例5:
本实施例提供了一种发酵工艺生产的药物及其制剂中十六烷基三甲基氯化铵的分离测定方法,所述方法除了气相色谱仪的条件为:毛细管柱中的固定液为甲基聚硅氧烷;
氮气为载气,控制载气流量为8mL/min;
气相色谱仪进样口温度为300℃;
气相色谱仪柱温为:在起始温度150℃下维持2min,再以10℃/min的速率升温至260℃,维持4min;
FID检测器的温度为320℃,其所通氢气的流量为50mL/min,所通空气的流量为500mL/min。
其他过程均与实施例1中相同,以外标法测定药物样品溶液中十六烷基三甲基氯化铵的含量为0.00301%,其回收率可达100.3%,精密度高,重复性良好。
对比例1:
本对比例提供了一种发酵工艺生产的药物及其制剂中十六烷基三甲基氯化铵的分离测定方法,所述方法除了气相色谱仪的条件中载气流量为15mL/min(>8mL/min)外,其他过程均与实施例1中相同,以外标法测定药物样品溶液中十六烷基三甲基氯化铵的含量为0.00415%,其准确率仅为138.3%,精密度较差。
对比例2:
本对比例提供了一种发酵工艺生产的药物及其制剂中十六烷基三甲基氯化铵的分离测定方法,所述方法除了气相色谱仪进样口温度为180℃(<250℃)外,其他过程均与实施例1中相同,以外标法测定药物样品溶液中十六烷基三甲基氯化铵的含量为0.00233%,其回收率仅为77.7%,精密度较差。
对比例3:
本对比例提供了一种发酵工艺生产的药物及其制剂中十六烷基三甲基氯化铵的分离测定方法,所述方法除了气相色谱仪进样口温度为350℃(>300℃)外,其他过程均与实施例1中相同,以外标法测定药物样品溶液中十六烷基三甲基氯化铵的含量为0.00432%,其回收率仅为144.0%,精密度较差。
对比例4:
本对比例提供了一种发酵工艺生产的药物及其制剂中十六烷基三甲基氯化铵的分离测定方法,所述方法除了气相色谱仪柱温起始温度为60℃(<90℃)外,其他过程均与实施例1中相同,以外标法测定药物样品溶液中十六烷基三甲基氯化铵的含量为0.00227%,其回收率仅为75.7%,精密度较差。
对比例5:
本对比例提供了一种发酵工艺生产的药物及其制剂中十六烷基三甲基氯化铵的分离测定方法,所述方法除了气相色谱仪柱温起始温度为200℃(>150℃)外,其他过程均与实施例1中相同,以外标法测定药物样品溶液中十六烷基三甲基氯化铵的含量为0.00251%,其回收率仅为83.7%,精密度较差。
对比例6:
本对比例提供了一种发酵工艺生产的药物及其制剂中十六烷基三甲基氯化铵的分离测定方法,所述方法除了气相色谱仪柱温终温温度为150℃(<240℃)外,其他过程均与实施例1中相同,以外标法测定药物样品溶液中十六烷基三甲基氯化铵的含量为0.00231%,其回收率仅为77.0%,精密度较差。
对比例7:
本对比例提供了一种发酵工艺生产的药物及其制剂中十六烷基三甲基氯化铵的分离测定方法,所述方法除了气相色谱仪柱温终温温度为300℃(>260℃)外,其他过程均与实施例1中相同,以外标法测定药物样品溶液中十六烷基三甲基氯化铵的含量为0.00219%,其回收率仅为73.0%,精密度较差。
综合实施例1-5和对比例1-6的结果可以看出,本发明所述分离测试方法通过合理浓度的样品溶液,优化气相色谱仪的工作参数,可有效避免药物中其他组成成分对于阳离子表面活性剂分离测试的干扰,进而可以将阳离子表面活性剂有效的从药物中分离出来,并准确算出药物中阳离子表面活性剂的含量,其回收率可达100%,精密度高,重复性好。且所用气相色谱仪与其他测试仪器相比,价格低廉,成本更低。
申请人声明,本发明通过上述实施例来说明本发明的详细方法,但本发明并不局限于上述详细方法,即不意味着本发明必须依赖上述详细方法才能实施。所属技术领域的技术人员应该明了,对本发明的任何改进,对本发明产品各原料的等效替换及辅助成分的添加、具体方式的选择等,均落在本发明的保护范围和公开范围之内。
- 还没有人留言评论。精彩留言会获得点赞!
- 一种阳离子型有机硅双子表面活性剂及其制备方法与流程
- 采用阴、阳离子复合表面活性剂的驱油方法与流程
- 一种双子阳离子表面活性剂及其制备方法和压裂液与流程
- 兽用阳离子表面活性剂复合消毒剂及其制备方法与应用与流程
- 包含有机硅烷、阳离子表面活性剂和具有大于或等于4meq/g电荷密度的阳离子聚合物的化妆品组合物的制造方法与工艺
- 一种阴、阳离子表面活性剂复配消毒剂技术的制造方法与工艺
- 一种含硒阳离子表面活性剂及其应用的制造方法与工艺
- 一种氧化胺表面活性剂及其制备方法和应用与制造工艺
- 双阳离子氟碳表面活性剂及其制备方法和作为双疏型润湿反转剂的应用和钻井液及其应用与制造工艺
- 一种葡萄糖基双子阳离子表面活性剂及其合成方法与制造工艺