一种西罗莫司的高效液相色谱检测方法与流程
本发明属于药物分析技术领域,具体涉及一种西罗莫司的高效液相色谱检测方法。
背景技术:
西罗莫司(sirolimus)又称雷帕霉素(rapamycin),是一种亲脂性含氮36元大环内酯类免疫抑制剂。1975年,加拿大Ayerst实验室的Vezina等人从太平洋Easler岛土壤样品中分离的吸水链霉菌(Streptomyees hygroscopicus)发酵液中首次得到。1977年西罗莫司被发现具有免疫抑制作用,1989年开始把西罗莫司作为治疗器官移植排斥反应的新药进行使用。1999年10月惠氏制药研制的西罗莫司口服溶液在美国首次上市,此后,1mg片剂也已在美上市。
国内西罗莫司产品主要有西罗莫司原料药、西罗莫司胶囊及西罗莫司口服溶液。目前,西罗莫司原料药及其制剂的有关物质与含量测定的现有检测技术主要是国家食品药品监督管理局标准(企业注册标准),具体包括原料药标准YBH09982005、YBH15302005、YBH02322008、YBH00972011和YBH01872012,胶囊标准YBH02472010,口服溶液标准YBH09992005、YBH14502005和YBH15312005。然而,各现有技术在重要参数质量控制上均存在显著缺陷,例如:
(1)流动相系统上,多项现有技术使用了甲醇,而西罗莫司在甲醇中存在结构转化现象,不适合作为溶解样品的溶剂或作为流动相的一部分。凡采用甲醇作为流动相组成的色谱系统,其供试品溶液西罗莫司主峰均存在和其前洗脱的杂质峰分离较差的现象,该现象也从侧面证实了以甲醇作为溶解样品的溶剂及作为流动相的一部分在有关物质控制上的局限性;
(2)流动相组成均为有机溶剂-水混合物,流动相系统不调节pH,不利于开环降解产物如断雷帕霉素等的控制,在此前提下采用等度洗脱方式,十分不利于弱极性强保留杂质的检测;
(3)在有关物质杂质的控制上,有2项现有技术提到已知杂质去甲基西罗莫司,1项现有技术提到去甲基西罗莫司和开环降解转化物,然而,其结论存在明显的错误,这是因为:在西罗莫司的检测过程中,所述开环降解转化物已被证实为断雷帕霉素与西罗莫司同系物及西罗莫司同分异构体的混合物,所述已知杂质去甲基西罗莫司已被证实为脯氨酰西罗莫司。
例如,中国专利申请CN105301159A披露了一种西罗莫司的高效液相色谱分析方法,其所采用的流动相为体积配比18:82~22:78的流动相A与流动相B的混合溶液,并且实施等度洗脱;其中,所述流动相A中的磷酸、庚烷磺酸钠、水的质量配比为0.1:0.01:1,所述流动相B中甲醇与乙腈的体积配比为15:85;由此可见,该专利申请未能避免甲醇的使用,且没有规避等度洗脱所带来的技术问题。
综上所述,现有技术在针对西罗莫司原料药及其制剂的有效可控的质量控制方面存在诸多缺陷与不足。因此,亟需提供一种全新的西罗莫司的分析方法或检测方法,在保证各相关杂质与有效成分高效分离的基础上,使其具有良好的专属性、重复性与准确度,从而更好地实现对西罗莫司的质量控制。
技术实现要素:
针对上述现有技术中存在的缺陷与不足,发明人旨在提供一种既具有高的分离度,又具有良好的专属性、重复性与准确度的分析方法或检测方法,用于西罗莫司原料药及其各种制剂的质量控制。
因此,本发明提供了一种西罗莫司的高效液相色谱检测方法,包括以下步骤:
将西罗莫司溶解于无水乙腈,制得西罗莫司样品溶液;取5~50μL进样至HPLC系统,该进样量与色谱柱载样量或检测器响应相适应;流速为1.0~2.5mL·min-1,具体操作时,采用与所选择的色谱柱尺寸相适应的流速;梯度洗脱,以277±2nm的检测波长进行HPLC分析;
其中,采用C18反相色谱柱,流动相由乙腈和缓冲盐溶液组成;所述缓冲盐溶液的pH为2.0~7.5,且所述缓冲盐溶液中盐的浓度为5~100mmol/L;
其中,乙腈占流动相总体积的体积百分比浓度为50~100%。
优选地,在上述西罗莫司的高效液相色谱检测方法中,所述西罗莫司包括西罗莫司原料药、西罗莫司片剂、西罗莫司胶囊或西罗莫司口服溶液。
优选地,在上述西罗莫司的高效液相色谱检测方法中,所述缓冲盐溶液的pH为3.5~4.0。
优选地,在上述西罗莫司的高效液相色谱检测方法中,所述缓冲盐溶液选自以下任一种体系:磷酸盐缓冲体系、甲酸铵缓冲体系、乙酸铵缓冲体系、三氟乙酸缓冲体系。所述缓冲盐溶液和质谱系统兼容,利于后续可实施的西罗莫司及其制剂中的杂质分析。
进一步优选地,在上述西罗莫司的高效液相色谱检测方法中,所述缓冲盐溶液为甲酸铵缓冲体系。
优选地,在上述西罗莫司的高效液相色谱检测方法中,所述缓冲盐溶液中盐的浓度为20~50mmol/L。
进一步优选地,在上述西罗莫司的高效液相色谱检测方法中,所述流动相由缓冲盐溶液、乙腈和改性剂组成。
更进一步优选地,在上述西罗莫司的高效液相色谱检测方法中,所述改性剂包括:甲基叔丁基醚和/或四氢呋喃。
更进一步优选地,在上述西罗莫司的高效液相色谱检测方法中,所述乙腈占流动相总体积的体积百分比浓度为50~99%,所述改性剂占流动相总体积的体积百分比浓度为1~5%。其中,所述改性剂的百分比浓度可根据西罗莫司的保留行为而做适当的调整。
综上所述,本发明所提供的西罗莫司的高效液相色谱检测方法,以无水乙腈为西罗莫司原料药及其制剂的溶剂,并以乙腈和缓冲盐溶液为流动相,可选地加入改性剂,意外地改善了脯氨酰西罗莫司、西罗莫司互变异构体A、去甲基西罗莫司及西罗莫司主峰之间的分离;其中,加入改性剂的主要目的和作用是调节西罗莫司及其杂质与固定相之间的相互作用力,最终目的是提高西罗莫司及其杂质之间的分离度,增加定量测定的准确度;从而避免了甲醇作为溶解样品的溶剂或作为流动相的一部分时,在有关物质控制上的局限性。本发明涉及的西罗莫司原料药及其制剂有关物质及含量测定的检测方法操作简便、专属性好,样品溶液稳定性好,可有效分离西罗莫司、互变异构体A、同系物及其工艺副产物(如脯氨酰西罗莫司、去甲基西罗莫司)、开环降解产物(如断雷帕霉素)等,检出的杂质个数较多,各相关色谱峰(工艺副产物及开环降解产物等)之间分离良好,准确度高且重复性好,可有效克服现有技术中存在的缺陷,从而有效控制西罗莫司原料药及其制剂的产品质量,亦可用于评估西罗莫司的生产工艺。
附图说明
图1~4依次为依据本发明实施例1所述的方法检测企业A、企业B、企业C和企业D的西罗莫司原料药所得到的典型液相色谱图;
图5为依据本发明实施例2所述的方法检测企业B的西罗莫司胶囊所得到的典型液相色谱图;
图6为依据现有国家食品药品监督管理局标准在甲醇-乙腈-水流动相体系中检测企业A的西罗莫司原料药所得到的典型液相色谱图;
图7为依据现有技术在甲醇-水流动相体系中检测企业A的西罗莫司原料药所得到的典型液相色谱图;
图8为依据现有技术在乙腈-水流动相体系中检测断雷帕霉素所得到的典型液相色谱图;
图9为依据本发明实施例3所述的方法检测企业A的西罗莫司原料药所得到的典型液相色谱图;
图10为依据本发明实施例4所述的方法检测企业A的西罗莫司胶囊所得到的典型液相色谱图;
图11为依据本发明实施例6所述的方法在磷酸盐缓冲体系中检测企业A的西罗莫司原料药所得到的典型液相色谱图;
图12为依据本发明实施例6所述的方法在乙酸铵缓冲体系中检测企业A的西罗莫司原料药所得到的典型液相色谱图;
图13为依据本发明实施例6所述的方法在三氟乙酸缓冲体系中检测企业A的西罗莫司原料药所得到的典型液相色谱图。
具体实施方式
下面结合具体实施方式对本发明作进一步阐述,但本发明并不限于以下实施方式。
本发明提供了一种西罗莫司的高效液相色谱检测方法,包括以下步骤:
将西罗莫司溶解于无水乙腈,制得西罗莫司样品溶液;取5~50μL进样至HPLC系统,流速为1.0~2.5mL·min-1,梯度洗脱,以277±2nm的检测波长进行HPLC分析;
其中,采用C18反相色谱柱,流动相由乙腈和缓冲盐溶液组成;所述缓冲盐溶液的pH为2.0~7.5,且所述缓冲盐溶液中盐的浓度为5~100mmol/L;
其中,乙腈占流动相总体积的体积百分比浓度为50~100%。
在一个优选实施例中,所述西罗莫司包括西罗莫司原料药、西罗莫司片剂、西罗莫司胶囊或西罗莫司口服溶液。
在一个优选实施例中,所述缓冲盐溶液的pH为3.5~4.0。
在一个优选实施例中,所述缓冲盐溶液选自以下任一种体系:磷酸盐缓冲体系、甲酸铵缓冲体系、乙酸铵缓冲体系、三氟乙酸缓冲体系。
在一个进一步优选的实施例中,所述缓冲盐溶液为甲酸铵缓冲体系。
在一个优选实施例中,所述缓冲盐溶液中盐的浓度为20~50mmol/L。
在一个进一步优选的实施例中,所述流动相由缓冲盐溶液、乙腈和改性剂组成。
在一个更进一步优选的实施例中,所述改性剂包括:甲基叔丁基醚和/或四氢呋喃。
在一个更进一步优选的实施例中,所述乙腈占流动相总体积的体积百分比浓度为50~99%,所述改性剂占流动相总体积的体积百分比浓度为1~5%。
以下实施例中采用的仪器为:Agilent 1200高效液相色谱仪,配备紫外检测器、四元梯度泵、自动进样器;电子天平(CP225D,德国Sartorius公司)。其中,高效液相色谱法所述使用的色谱柱为Kromasil C18(4.6mm×250mm,5μm)色谱柱;检测波长为277nm;流速:1.5mL·min-1,进样体积:20μL。
实施例1
HPLC检测
西罗莫司样品溶液的制备:取西罗莫司原料药,用无水乙腈溶解,配制成1mg/mL的溶液;取20μL西罗莫司样品溶液,进样,对西罗莫司原料药中的主要成分、有关物质和杂质进行定性和定量分析;
其中,流动相A为20mM甲酸铵缓冲溶液(即甲酸铵缓冲体系,pH约为3.8),流动相B为乙腈-甲基叔丁基醚(体积比99:1,下文中的百分比同样表示体积百分比),其中甲基叔丁基醚作为改性剂;线性梯度洗脱条件为:0~18min:43%A、57%B;18~25min:29%A、71%B;25~33min:100%B;33~43min:100%B(维持10分钟);43min后:43%A、57%B;
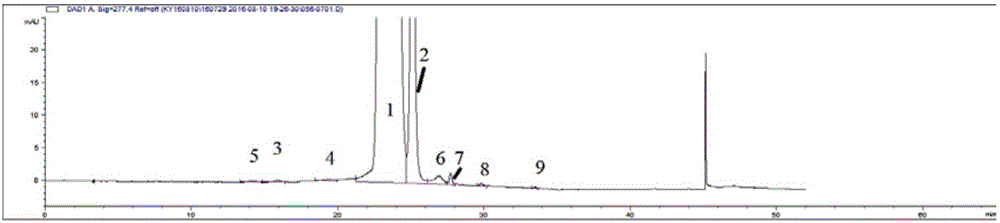
分别来自企业A、企业B、企业C和企业D的西罗莫司原料药(不同来源的)的检测结果见图1~4。在图1~4中,色谱峰1为西罗莫司,2为西罗莫司互变异构体C,其余色谱峰为主要杂质峰:3为脯氨酰西罗莫司,4为去甲基西罗莫司。
此实施例1的检测结果表明,不同来源的西罗莫司原料药之间的杂质谱存在较大差异;并且,西罗莫司主峰及其杂质峰得到了良好的分离。
对比例1
此外,发明人还依据现有国家食品药品监督管理局标准对企业A的西罗莫司原料药进行了HPLC分析,其中采用甲醇-乙腈-水作为流动相,获得了如图6所示的液相色谱图;如图6所示,西罗莫司主峰1和其相邻的主要工艺杂质——脯氨酰西罗莫司(杂质峰3)和去甲基西罗莫司(杂质峰4)不能得到有效分离。
进一步地,发明人按照实施例1中的步骤制备了源自企业A的西罗莫司原料药的西罗莫司样品溶液,并以相同的液相条件进行了HPLC分析,唯一的区别在于使用了甲醇作为流动相B,使用了水作为流动相A,该HPLC分析所获得的液相色谱图为图7。如图7所示,在采用甲醇-水作为流动相的条件下,西罗莫司主峰1前的多种杂质不能得到有效分离。
因此,发明人得出结论,无论是甲醇-乙腈-水流动相体系还是甲醇-水流动相体系,都会影响HPLC检测中的分离效果,均难以准确反映西罗莫司的纯度,进而难以获得有效的质量控制。
通过比较实施例1与对比例1,可发现本发明所建立的色谱分离系统,对西罗莫司和其工艺杂质及开环降解产物之间的分离良好,特别是脯氨酰西罗莫司与去甲基西罗莫司以及去甲基西罗莫司和西罗莫司之间的分离均较好,可以分别准确的控制主要工艺杂质的含量,同时有效反映西罗莫司的质量,从而可为企业生产工艺的监控提供技术保障;与此同时,本发明所提供的西罗莫司的高效液相色谱检测方法,通过对西罗莫司产品的质量控制,能够有效区分不同的西罗莫司生产工艺的优劣。由此可见,避免甲醇作为溶解样品的溶剂或作为流动相的一部分,是本发明所述西罗莫司的高效液相色谱检测方法取得优异技术效果(主要是分离效果)的关键因素之一。
实施例2
HPLC检测
西罗莫司样品溶液的制备:取来自企业B的西罗莫司胶囊,用无水乙腈溶解,配制成0.5mg/mL的溶液;取20μL西罗莫司样品溶液,进样,对西罗莫司胶囊中的主要成分、有关物质和杂质进行定性和定量分析;
其中,流动相A为20mM甲酸铵缓冲溶液(即甲酸铵缓冲体系,pH约为3.8),流动相B为乙腈-甲基叔丁基醚(体积比99:1,下文中的百分比同样表示体积百分比),其中甲基叔丁基醚作为改性剂;线性梯度洗脱条件为:0~18min:43%A、57%B;18~25min:29%A、71%B;25~33min:100%B;33~43min:100%B(维持10分钟);43min后:43%A、57%B;
检测结果见图5,其中:色谱峰1为西罗莫司,2为西罗莫司互变异构体C,其余色谱峰为主要杂质峰:其中4为去甲基西罗莫司,6为西罗莫司同系物,8为氧化物类似物,11为氧化聚合物类似物,12为西罗莫司互变异构体A(由西罗莫司和其互变异构体C相互转化过程中产生,随着样品放置时间的增加含量增加;并且制剂过程也可产生),13为断雷帕霉素。
值得说明的是,断雷帕霉素为西罗莫司的开环降解产物,通常在制剂过程或贮藏中产生,其作为制剂中表征降解产物的指针性杂质,是反映产品内在质量的主要考察指标之一。另外,所述断雷帕霉素结构中含有羧基,因此,在乙腈-水流动相体系中断雷帕霉素为解离状态,HPLC检测断雷帕霉素时(液相条件和操作,与此实施例2中对西罗莫司胶囊检测的液相条件和操作相同,唯一区别在于将西罗莫司胶囊替换为断雷帕霉素标准品),不被保留,且在死时间区域洗脱,如图8所示。因此,在乙腈-水流动相体系中,对断雷帕霉素的质量控制无法得以实现;对于胶囊等制剂来说,采用乙腈-水流动相体系的方法无法反映生产过程中的制剂工艺及贮藏过程对西罗莫司最终产品质量的影响。
由于本发明所建立的色谱分离系统的水相为一定pH值的缓冲溶液,这不仅有利于保障西罗莫司和其工艺杂质的分离,而且能够增加主要开环降解产物——断雷帕霉素的保留,同时确保降解产物和工艺杂质之间的分离维持良好,从而能够有效反映并监控制剂过程及贮藏过程中西罗莫司制剂的质量。
实施例3
HPLC检测
西罗莫司样品溶液的制备:取来自企业A的西罗莫司原料药,用无水乙腈溶解,配制成1mg/mL的溶液;取20μL西罗莫司样品溶液,进样,对西罗莫司胶囊中的主要成分、有关物质和杂质进行定性和定量分析;
其中,流动相A为20mM甲酸铵缓冲溶液(即甲酸铵缓冲体系,pH约为3.8),流动相B为乙腈-四氢呋喃(体积比97:3),其中四氢呋喃作为改性剂;线性梯度洗脱条件为:0~20min:44%A、56%B;20~30min:57%A、43%B;30~40min:100%B;40~50min:100%B(维持10分钟);50min后:44%A、56%B;
检测结果见图9,其中:色谱峰1为西罗莫司,2为西罗莫司互变异构体C,其余色谱峰为主要杂质峰:其中3为脯氨酰西罗莫司,4为去甲基西罗莫司。
实施例4
HPLC检测
西罗莫司样品溶液的制备:取来自企业A的西罗莫司胶囊,用无水乙腈溶解,配制成0.5mg/mL的溶液;取20μL西罗莫司样品溶液,进样,对西罗莫司胶囊中的主要成分、有关物质和杂质进行定性和定量分析;
其中,流动相A为20mM甲酸铵缓冲溶液(pH约为3.8),流动相B为乙腈-四氢呋喃(体积比97:3),其中四氢呋喃作为改性剂;线性梯度洗脱条件为:0~20min:44%A、56%B;20~30min:57%A、43%B;30~40min:100%B;40~50min:100%B(维持10分钟);50min后:44%A、56%B;
检测结果见图10,其中:色谱峰1为西罗莫司,2为西罗莫司互变异构体C,其余色谱峰为主要杂质峰:其中4为去甲基西罗莫司,6为西罗莫司同系物,8为氧化物类似物,12为西罗莫司互变异构体A(由西罗莫司和其互变异构体C相互转化过程中产生,随着样品放置时间的增加含量增加;并且制剂过程也可产生),13为断雷帕霉素。
实施例5
此外,发明人考察了改性剂对西罗莫司有关物质色谱峰分离效果的影响。
具体地,发明人按照实施例1中的检测条件与操作步骤分别考察了不同体积百分比浓度的甲基叔丁基醚作为改性剂时,西罗莫司与其相关杂质之间的分离情况(参见下表1);发明人按照实施例3中的检测条件与操作步骤分别考察了不同体积百分比浓度的四氢呋喃作为改性剂时,西罗莫司与其相关杂质之间的分离情况(参见下表2)。
表1
表2
由于西罗莫司结构式中含有多个含氧极性基团,西罗莫司主要的生产工艺杂质为其结构类似物,简单的常规流动相体系如甲醇体系、乙腈体系较难实现西罗莫司与其生产工艺杂质间的优良分离。并且,西罗莫司在溶液中存在化学结构相互转化的现象,这更增加了色谱分离的难度。
发明人在研究中意外地发现,将甲基叔丁基醚和四氢呋喃掺入流动相均可以调节西罗莫司及其相关杂质和流动相之间的相互作用力,进而改善结构相似的各种杂质之间以及各种杂质和西罗莫司主峰之间的分离。
如表1所示,随着甲基叔丁基醚添加量的增加,脯氨酰西罗莫司和西罗莫司互变异构体A分离度显著增加,西罗莫司互变异构体A和去甲基西罗莫司间分离度未见显著增加;且随着甲基叔丁基醚添加量的增加,西罗莫司和互变异构体间的分离难以通过乙腈比例的调节来改善,甲基叔丁基醚添加量过大时甚至导致了西罗莫司及其互变异构体之间的分离度更差;因此,表1中的数据说明甲基叔丁基醚和西罗莫司及其相关物质(各种杂质)之间的相互作用力均比较显著,从而宜采用低的改性剂添加比例,即采用较少的甲基叔丁基醚添加量。
如表2所示,随着四氢呋喃添加量的增加,脯氨酰西罗莫司和西罗莫司互变异构体A分离度显著增加,西罗莫司互变异构体A和去甲基西罗莫司之间的分离度也呈现增加趋势,并且可以通过调整流动相体系中乙腈的比例来调整西罗莫司的保留,且可以保证各相关检测物之间良好的分离,例如,西罗莫司及其互变异构体之间的分离状况良好;因此,表2中的数据说明四氢呋喃和西罗莫司及其相关物质(各种杂质)之间的相互作用力不同,因而,可通过乙腈比例的调节以调整西罗莫司的保留,并可通过适当提升四氢呋喃添加量来改善西罗莫司及其相关物质之间的分离,因此,四氢呋喃用作改性剂时的效果优于甲基叔丁基醚;换言之,本发明所述的西罗莫司的高效液相色谱检测方法更优选四氢呋喃作为改性剂。
由此可知,发明人综合考虑了脯氨酰西罗莫司、西罗莫司互变异构体A与去甲基西罗莫司三者之间的分离度,并在西罗莫司主峰的保留时间符合要求的前提下,本发明实施例1和2中以含改性剂甲基叔丁基醚为例进行阐述,所使用的甲基叔丁基醚的添加量约为1%;本发明实施例3和4中以含改性剂四氢呋喃为例进行阐述,所使用的四氢呋喃的添加量约为2%。
实施例6
此外,发明人考察了不同缓冲盐溶液种类对西罗莫司有关物质色谱峰分离效果的影响。
具体地,发明人按照实施例1中的检测条件与操作步骤分别考察了采用不同缓冲体系时(磷酸盐缓冲体系、乙酸铵缓冲体系、三氟乙酸缓冲体系),西罗莫司与其相关杂质之间的分离情况,即对西罗莫司有关物质色谱峰保留行为的影响。如图11~13所示,其中,图11显示的来自企业A的西罗莫司原料药的液相色谱图对应磷酸盐缓冲体系,图12显示的来自企业A的西罗莫司原料药的液相色谱图对应乙酸铵缓冲体系,图13显示的来自企业A的西罗莫司原料药的液相色谱图对应三氟乙酸缓冲体系;其中,西罗莫司主峰在各缓冲体系中均能够与其相关杂质(主要为工艺杂质)之间分离良好。发明人通过分析认为,这一结果的主要原因为:西罗莫司及其工艺杂质均为近中性化合物,不含有可解离基团,因此对于不同的缓冲体系中未表现出明显的分离差异。
对于西罗莫司的主要降解产物——断雷帕霉素,由于其结构式中含有羧基,其保留行为易受流动相pH的影响。发明人在研究过程中发现,流动相pH值在3.5左右时,可实现断雷帕霉素和工艺杂质(如脯氨酰西罗莫司、去甲基西罗莫司)之间的良好分离;考虑到各酸的pKa值及相应的缓冲范围,并且为了通过可后续实施的质谱检测等技术手段更好地实现对杂质谱的系统分析,更优选权利要求1和2中所采用的甲酸铵缓冲体系,作为缓冲体系(缓冲盐溶液)。
实施例7
另外,发明人考察了缓冲盐溶液的不同浓度对西罗莫司有关物质色谱峰分离效果的影响。
具体地,发明人按照实施例1中的检测条件与操作步骤分别考察了甲酸铵的浓度为5、10、20、25、50、100mmol/L的甲酸铵缓冲体系(甲酸铵溶液),对西罗莫司与其相关杂质之间的分离情况的影响,即对西罗莫司有关物质色谱峰保留行为的影响。
检测结果表明,随着甲酸铵缓冲体系中甲酸铵浓度的增加,即随着缓冲盐溶液中盐的浓度的增加,西罗莫司及其相关杂质之间的保留均减弱,但对各色谱峰之间的分离度未见显著影响。发明人通过分析,认为其原因主要为:西罗莫司及其工艺杂质均为近中性化合物,不含有可解离基团,因此对于不同缓冲容量的甲酸铵缓冲溶液,仅表现为缓冲盐离子强度差异所导致的色谱峰保留强弱略有不同,而并没有对分离度产生显著影响。综合考虑缓冲体系的缓冲容量及各色谱峰的保留行为,优选所述缓冲盐溶液中盐的浓度为20~50mmol/L。
以上对本发明的具体实施例进行了详细描述,但其只是作为范例,本发明并不限于以上描述的具体实施例。对于本领域技术人员而言,任何对本发明进行的等同修改和替代也都在本发明的范畴之中。因此,在不脱离本发明的精神和范围下所作的均等变换和修改,都应涵盖在本发明的范围内。
- 还没有人留言评论。精彩留言会获得点赞!