用高效液相色谱法测定盐酸屈他维林注射液中有关物质的方法与流程
本发明涉及药物分析
技术领域:
,尤其是涉及一种用高效液相色谱法测定盐酸屈他维林注射液中有关物质的方法。
背景技术:
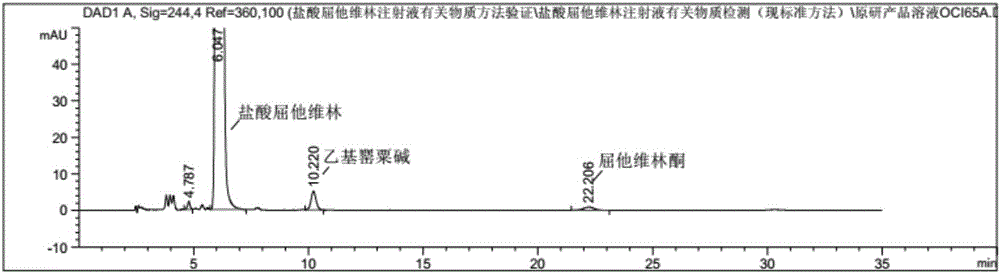
:盐酸屈他维林(Drotaverinehydrochloride),化学名称为:1-{(3,4-二乙氧基苯基)亚甲基}-6,7-二乙氧基-1,2,3,4-四氢异喹啉盐酸盐,商品名为诺仕帕(RNO-SPA),是由法国赛诺菲公司在匈牙利的合资公司Chinoin药厂研制开发的一种新型解痉挛药。该药是异喹啉类衍生物,与传统山莨菪碱,阿托品等抗胆碱能解除痉挛药不同,它直接作用于平滑肌细胞,其舒张平滑肌的作用机制为:作用于平滑肌细胞表面,改变细胞膜电位和通透性;抑制磷酸二酯酶,增加肌细胞内环磷腺苷(cAMP)水平;抑制细胞内最初的钙离子反应,平滑肌舒张,解除痉挛。其特点是:(1)作用全面,疗效持久,盐酸屈他维林对平滑肌痉挛性收缩的每一环节都起作用,特别是高张力平滑肌有特效,其消除半衰期为9-11h,故有效期长;(2)不良反应小,盐酸屈他维林不影响自主神经系统,对胃肠道正常的平滑肌无影响,没有M受体拮抗剂的不良反应。该药的适应症包括冠脉功能不全、闭塞性动脉内膜炎、心绞痛、胃肠道平滑肌痉挛、肠易激综合征、胆绞痛、肾绞痛和泌尿道痉挛、子宫痉挛、痛经等。其优点在于无严重心血管反应、无副交感神经阻滞、无瞳孔放大、无青光眼的急性发作、无尿道膀胱的扩张,起效快,不良反应少。盐酸屈他维林原料药的成分是明确的,主要包含盐酸屈他维林及2种相关杂质(乙基罂粟碱、屈他维林酮),将这2种物质彻底的快速的分离十分重要。盐酸屈他维林注射液国家局标准(YBH17602006)有关物质高效液相分析方法参照盐酸屈他维林注射液复核标准JX20000051、盐酸屈他维林片质量标准WS-2127(X-184)-2002制定。国家局标准(YBH17602006),采用磷酸盐缓冲液(pH3.3±0.05)-乙腈-甲醇等度洗脱,分析时间周期较短,主峰约5~7分钟出峰,有后拖尾现象,可能存在未知杂质与主峰未有效分离的情况,各已知杂质分离较好,但盐酸屈他维林注射液经提高灭菌工艺条件,有3个未知杂质降解明显,分离不理想,在保留时间约3~5分钟之间某些杂质相互重叠,测定不准确,不能达到有关物质检查要求,不利于实际推广应用。因此,需要对盐酸屈他维林注射液有关物质分析方法进行改进,建立一个快速、有效的检测方法,在灭菌工艺由100℃灭菌20分钟,变为121℃灭菌15分钟后,能区分并检测出盐酸屈他维林原料药内的所有相关物质,有利于提高盐酸屈他维林注射液的产品质量,提高患者用药安全性。针对上述问题,特提出本发明。技术实现要素:本发明的目的是探索盐酸屈他维林有关物质高效液相色谱分析的各种条件,提供一个简便、可靠的盐酸屈他维林有关物质高效液相色谱分析方法。为解决上述技术问题,本发明采用如下技术方案:一种用高效液相色谱法测定盐酸屈他维林注射液中有关物质的方法,该方法包括如下步骤:(1)配制供试样品溶液:取盐酸屈他维林用溶剂配制成供试样品溶液;(2)配制对照品溶液:取盐酸屈他维林、乙基罂粟碱和屈他维林酮用溶剂配制成对照品溶液;(3)配制系统适用性溶液:取盐酸屈他维林、乙基罂粟碱和屈他维林酮用溶剂配制成系统适用性溶液;(4)进行系统适用性实验;(5)进行检测灵敏度实验;用对照品溶液调节高效液相色谱仪的检测灵敏度,使主成分色谱峰的峰高为满量程的10-25%;(6)按照下列高效液相色谱条件进行测定:流动相:以磷酸盐缓冲液作为流动相A,以乙腈为流动相B,以甲醇为流动相C;所述磷酸盐缓冲液为0.1%磷酸水溶液用三乙胺调节至pH=3.3±0.05;紫外检测器:检测波长为200-400nm;色谱柱:以十八烷基硅烷键合硅胶为填充剂;流速:0.5-2.0mL/min;柱温:20-60℃;(7)按外标法,计算有关物质的含量。优选地,所述的紫外检测器的检测波长为200-300nm,更优选的为244nm。优选地,所述流速为1.0mL/min;所述柱温为30-50℃,更优选地为40℃。优选地,所述磷酸盐缓冲液由0.1%磷酸水溶液用三乙胺调节pH3.3±0.05后制成。优选地,所述供试样品溶液的配制具体包括配制含盐酸屈他维林0.8mg/mL的溶液,所述溶液的溶剂为流动相A、甲醇和乙腈形成的混合溶剂,所述混合溶剂为将甲醇和乙腈按体积比2:1混合后的混合液再与流动相A按体积比1:9混合。优选地,所述对照品溶液的配制具体包括配制含盐酸屈他维林、乙基罂粟碱和屈他维林酮的溶液,所述溶液中盐酸屈他维林的浓度为4.0μg/mL;乙基罂粟碱的浓度为1.6μg/mL;屈他维林酮的浓度为4.0μg/mL;将盐酸屈他维林、乙基罂粟碱和屈他维林酮先用甲醇与乙腈体积比为2:1的混合液溶解,然后加入流动相A、甲醇和乙腈形成的混合溶剂,所述混合溶剂为将甲醇和乙腈按体积比2:1混合后的混合液再与流动相A按体积比1:9混合。优选地,所述系统适用性溶液的配制具体包括配制含盐酸屈他维林、乙基罂粟碱和屈他维林酮的溶液,所述溶液中盐酸屈他维林的浓度为0.8mg/mL;乙基罂粟碱的浓度为1.6μg/mL;屈他维林酮的浓度为4.0μg/mL;将盐酸屈他维林、乙基罂粟碱和屈他维林酮先用甲醇与乙腈体积比为2:1的混合液溶解,然后加入流动相A、甲醇和乙腈形成的混合溶剂,所述混合溶剂为将甲醇和乙腈按体积比2:1混合后的混合液再与流动相A按体积比1:9混合。优选地,所述系统适用性试验是指取系统适用性溶液20μL注入液相色谱仪,盐酸屈他维林与乙基罂粟碱的分离度大于3.0,各杂质峰之间分离度符合要求,理论塔板数按盐酸屈他维林计算,不低于3000。优选地,以流动相A、流动相B和流动相C进行梯度洗脱;所述的梯度为:本发明的有益效果如下:本发明立足于高效液相色谱分析方法,针对现有技术存在的分离不彻底、峰形拖尾严重、包峰等缺点,采用新的缓冲液体系以及流动相比例,使杂质能够完全分离,解决了现有技术的相关问题;且本发明提供的方法经专属性、灵敏度等方法学研究及验证,其验证结果均在可接受范围;本发明同现行标准进行专属性对比研究,证明其方法更能有效地控制盐酸屈他维林注射液有关物质;本发明方法还具有下述有益效果,1.流动相配制较为简单易操作;2.能够有效分离杂质,主成分无拖尾,检出的杂质种类、数量均高于国家局标准(YBH17602006)方法,检测准确度高,提高产品的安全性,方法科学、可靠,可控;3.操作简便,适于实际应用和推广。附图说明图1示出了本申请比较例1提供的检测结果的HPLC图谱;图2示出了本申请比较例2提供的检测结果的HPLC图谱;图3示出了本申请实施例1提供的检测结果的HPLC图谱;图4示出了本申请实施例2提供的检测结果的HPLC图谱;图5示出了本申请实施例3提供的检测结果的HPLC图谱;图6A示出了本申请比较例3提供的检测结果的HPLC图谱;图6B示出了本申请实施例4提供的检测结果的HPLC图谱;图7A示出了本申请比较例4提供的检测结果的HPLC图谱;图7B示出了本申请实施例5提供的检测结果的HPLC图谱;图8A示出了本申请比较例5提供的检测结果的HPLC图谱;图8B示出了本申请实施例6提供的检测结果的HPLC图谱;图9A示出了本申请比较例6提供的检测结果的HPLC图谱;图9B示出了本申请实施例7提供的检测结果的HPLC图谱;图10A示出了本申请比较例7提供的检测结果的HPLC图谱;图10B示出了本申请实施例8提供的检测结果的HPLC图谱;图11示出了本申请实施例14提供的检测结果的HPLC图谱;图12示出了本申请实施例14提供的检测结果的HPLC图谱;图13示出了本申请实施例14提供的检测结果的HPLC图谱;图14示出了本申请实施例14提供的检测结果的HPLC图谱。具体实施方式下面将结合附图对本发明的技术方案进行清楚、完整地描述,显然,所描述的实施例是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。根据盐酸屈他维林原料合成工艺路线及试验,可以确定可能的有关物质为:(1)起始物料:3,4-二乙氧基苯乙酸和3,4-二乙氧基苯乙胺;(2)合成中间体(缩合物):N-(3,4-二乙氧基苯乙酰)-β-(3,4-二乙氧基苯)乙胺;(3)降解产物:乙基罂粟碱、屈他维林酮。起始物料和中间体在原料制备工艺中已经加以控制,并制定内控标准。本方法重点关注2个已知降解产物乙基罂粟碱、屈他维林酮和灭菌过程中降解变大的3个未知杂质。步骤1.配制供试样品溶液:精密量取盐酸屈他维林注射液适量,加溶剂2定量稀释制成每1mL中约含盐酸屈他维林0.8mg的溶液,作为供试品溶液;步骤2.配制对照品溶液:精密称取盐酸屈他维林、乙基罂粟碱与屈他维林酮各适量,加溶剂1适量溶解,再用溶剂2稀释制成每1mL约含盐酸屈他维林4.0μg、乙基罂粟碱1.6μg与屈他维林酮4.0μg的混合溶液,作为对照品溶液;步骤3.配制系统适用性溶液:精密称取盐酸屈他维林、乙基罂粟碱与屈他维林酮各适量,加溶剂1适量溶解,再用溶剂2稀释成每1mL约含乙基罂粟碱1.6μg、屈他维林酮4.0μg与盐酸屈他维林0.8mg的混合溶液,作为系统适用性溶液;步骤4.检测:以十八烷基硅烷键合硅胶作为色谱柱填料,以磷酸盐缓冲液(0.1%磷酸水溶液用三乙胺调节pH3.3±0.05)作为流动相A、以乙腈作为流动相B,以甲醇作为流动相C,采用梯度洗脱法,检测所述供试样品溶液,检测波长为244nm;柱温为40℃,流速为1.0mL/分钟。梯度条件见表1;表1步骤5.精密量取系统适用性溶液20μL注入液相色谱仪,记录色谱图。理论板数按盐酸屈他维林计算,应不低于3000,盐酸屈他维林峰与乙基罂粟碱的分离度应大于3.0,各杂质峰之间分离度符合要求;步骤6.精密量取对照品溶液20μL,注入液相色谱仪,调节检测灵敏度,使主成分色谱峰的峰高为满量程的10%-25%;步骤7.按外标法,计算乙基罂粟碱、屈他维林酮、其他单个杂质的含量;上述步骤1、2和3中,所述溶剂1为甲醇乙腈混合溶液(甲醇︰乙腈=2︰1);溶剂2为流动相A︰甲醇乙腈混合溶液(甲醇︰乙腈=2︰1)=9︰1。以下实施例和比较例中使用的实验仪器均为安捷伦高效液相色谱仪;使用的色谱柱为COSMOSIL5C18-MS-Ⅱ250mm×4.6mm5μm;各实施例中,以磷酸盐缓冲液(0.1%磷酸水溶液用三乙胺调节pH3.3±0.05)作为流动相A、以乙腈作为流动相B,以甲醇作为流动相C,溶剂1为甲醇乙腈混合溶液(甲醇︰乙腈=2︰1);溶剂2为流动相A︰甲醇乙腈混合溶液(甲醇︰乙腈=2︰1)=9︰1;检测所述供试样品溶液,检测波长为244nm;柱温为40℃,流速为1.0mL/分钟,进样量:20μL。梯度洗脱按照表2进行线性洗脱:表2杂质信息见下表3:表3杂质信息汇总实施例1-3系统适用性试验实施例1,系统适用性溶液配制:分别取盐酸屈他维林、已知杂质屈他维林酮、已知杂质乙基罂粟碱各适量,加溶剂1适量溶解,在用溶剂2稀释制成每1mL约含屈他维林酮4.0μg(0.5%)、乙基罂粟碱1.6μg(0.2%)、盐酸屈他维林0.8mg的混合溶液,作为系统适用性溶液;实施例2,供试品溶液配制:精密量取盐酸屈他维林注射液适量,加溶剂2定量稀释制成每1mL中约含盐酸屈他维林0.8mg的溶液,作为供试品溶液;实施例3,选择性溶液配制:分别取盐酸屈他维林、已知杂质屈他维林酮、已知杂质乙基罂粟碱、屈他维林缩合物(中间体)、起始物料3,4-二乙氧基苯乙酸和3,4-二乙氧基苯乙胺各适量加溶剂1适量溶解,再用溶剂2稀释制成每1mL约含屈他维林酮、屈他维林缩合物、3,4-二乙氧基苯乙酸、3,4-二乙氧基苯乙胺各4.0μg(0.5%)、乙基罂粟碱1.6μg(0.2%)、盐酸屈他维林0.8mg的混合溶液,作为选择性溶液;进样并记录色谱图。比较例1-2系统适用性试验按盐酸屈他维林注射液质量标准YBH17602006试验;色谱条件:色谱柱:十八烷基硅烷键合硅胶为填充剂;流动相:磷酸盐缓冲液(取磷酸氢二铵13.2g,加水200mL使溶解,加85%的磷酸7.6mL,用水稀释至1000mL,调节pH至3.3±0.05)-乙腈-甲醇(800∶400∶800);溶剂:0.1mol/LHCl-甲醇-乙腈(8∶1∶1);流速:1.0mL/min;波长:244nm;柱温:40℃;进样量:20μL;比较例1,系统适用性溶液配制:分别取盐酸屈他维林、已知杂质屈他维林酮、已知杂质乙基罂粟碱各适量加溶剂溶解稀释制成每1mL约含屈他维林酮4.0μg(0.5%)、乙基罂粟碱1.6μg(0.2%)、盐酸屈他维林0.8mg的混合溶液,作为系统适用性溶液;比较2,供试品溶液配制:精密量取盐酸屈他维林注射液适量,加溶剂定量稀释制成每1mL中约含盐酸屈他维林0.8mg的溶液,作为供试品溶液;进样并记录色谱图。结论:比较例1和比较例2见图1,图2所示:系统适用性溶液中,乙基罂粟碱、屈他维林酮和主成分分离度较好,但主峰保留时间较短,有拖尾现象,存在未知杂质与主峰未有效分离的情况;另外,溶剂峰后杂质出峰时间较快,且未知杂质分离不理想,在保留时间约3~5分钟之间部分杂质相互重叠,分离效果不能达到有关物质检查要求。实施例1-3结果见图3、图4所示:乙基罂粟碱、屈他维林酮和主成分分离度较好,且主成分保留时间适中,无拖尾现象;另外,溶剂峰后各未知杂质分离效果较好;图5所示:选择性溶液中,加入起始物料、中间体、已知杂质考察分离度,结果显示,盐酸屈他维林与3,4-二乙氧基苯乙酸、3,4-二乙氧基苯乙胺、屈他维林缩合物、乙基罂粟碱、屈他维林酮5个杂质的分离度均大于3.0,分离效果能达到有关物质检查要求。实施例4-8破坏性试验供试品溶液制备实施例4,破坏前溶液:取盐酸屈他维林注射液1mL(约相当于盐酸屈他维林20mg)置于25mL容量瓶中,加溶剂2稀释至刻度,摇匀,作为未破坏溶液。实施例5,高温破坏溶液:取盐酸屈他维林注射液1mL(约相当于盐酸屈他维林20mg)置于25mL容量瓶中,100℃加热8h,冷却至室温,溶剂2稀释至刻度,摇匀,作为高温破坏溶液。实施例6,光照破坏溶液:取盐酸屈他维林注射液1mL(约相当于盐酸屈他维林20mg),置于25mL容量瓶中,紫外灯下光照15h,溶剂2稀释至刻度,作为光照破坏溶液。实施例7,碱破坏溶液:取盐酸屈他维林注射液1mL(相当于盐酸屈他维林约20mg),置于25mL容量瓶中,加1mol/L氢氧化钠1mL,60℃破坏1.5h后,加1mol/L盐酸1mL中和,溶剂2稀释至刻度,摇匀,作为碱破坏溶液。实施例8,氧化破坏溶液:取盐酸屈他维林注射液1mL(约相当于盐酸屈他维林20mg),置于25mL容量瓶中,加30%的双氧水2mL,60℃破坏8h,溶剂2稀释至刻度,摇匀,作为氧化破坏溶液。进样并记录色谱图。比较例3-7破坏性试验按盐酸屈他维林注射液质量标准YBH17602006实验,色谱条件:色谱柱:十八烷基硅烷键合硅胶为填充剂;流动相:磷酸盐缓冲液(取磷酸氢二铵13.2g,加水200mL使溶解,加85%的磷酸7.6mL,用水稀释至1000mL,调节pH至3.3±0.05)-乙腈-甲醇(800∶400∶800);溶剂:0.1mol/LHCl溶液-甲醇-乙腈(8∶1∶1);流速:1.0mL/min;波长:244nm;柱温:40℃;进样量:20μL;供试品溶液制备:比较例3,破坏前溶液:取盐酸屈他维林注射液1mL(约相当于盐酸屈他维林20mg)置于25mL容量瓶中,加溶剂稀释至刻度,摇匀,作为未破坏溶液;比较例4,高温破坏溶液:取盐酸屈他维林注射液1mL(约相当于盐酸屈他维林20mg)置于25mL容量瓶中,100℃加热8h,冷却至室温,溶剂稀释至刻度,摇匀,作为高温破坏溶液。比较例5,光照破坏溶液:取盐酸屈他维林注射液1mL(约相当于盐酸屈他维林20mg),置于25mL容量瓶中,紫外灯下光照15h,溶剂稀释至刻度,作为光照破坏溶液。比较例6,碱破坏溶液:取盐酸屈他维林注射液1mL(相当于盐酸屈他维林约20mg),置于25mL容量瓶中,加1mol/L氢氧化钠1mL,60℃破坏1.5h后,加1mol/L盐酸1mL中和,溶剂稀释至刻度,摇匀,作为碱破坏溶液。比较例7,氧化破坏溶液:取盐酸屈他维林注射液1mL(约相当于盐酸屈他维林20mg),置于25mL容量瓶中,加30%的双氧水2mL,60℃破坏8h,溶剂稀释至刻度,摇匀,作为氧化破坏溶液。结论:比较例3-7结果见图6A,7A,8A,9A,10A所示:各降解条件下,除已知杂质乙基罂粟碱和屈他维林酮外,其它降解杂质均在主峰出峰时间(RT=6.1)之前,且在保留时间约3~5分钟之间有重叠,分离度较差,影响检测结果。实施例4-8结果见图6B,7B,8B,9B,10B所示:可以得知(1)供试品在各种破坏条件下的降解产物保留时间均大于3.0分钟,各破坏降解产物与溶剂峰及主峰均能很好分离,溶剂不干扰有关物质测定;各杂质峰分离度均大于3.0;(2)盐酸屈他维林注射液经破坏性试验,与未破坏相对比,乙基罂粟碱:高温、碱和氧化破坏条件下含量变化较大,光照破坏未见明显变化;屈他维林酮:碱破坏含量变化明显,高温破坏和光照破坏含量变化较大,氧化破坏未见明显变化;未知杂质:均有不同程度降解产生,光照、氧化较为明显;(3)除高温破坏溶液外,其他破坏溶液和破坏前溶液杂质个数均比国家局标准(YBH17602006)方法检查杂质数多,屈他维林酮含量、最大单杂含量和杂质总量均高于国家局标准标准检查方法结果。综上所述,本发明有关物质检查方法测得破坏前溶液和破坏溶液杂质数,屈他维林酮含量、其他最大单杂含量和杂质总量均高于国家局标准(YBH17602006)检查方法结果。因此,修改后有关物质检查方法的专属性和分离效能优于现标准检查方法。以下为本发明盐酸屈他维林注射液有关物质检查方法验证:实施例9检测限和定量限取专属性项下盐酸屈他维林(浓度:4μg/mL)、乙基罂粟碱(浓度:1.6μg/mL)、屈他维林酮(浓度:4μg/mL)的单一定位溶液,用稀释法测其定量限(S/N≥10)、检测限(S/N≥3),结果见表4表4检测限和定量限结论:本色谱条件下,盐酸屈他维林、乙基罂粟碱和屈他维林酮的定量限与检测限均符合接受限度。实施例10标准曲线及线性范围精密称取盐酸屈他维林、屈他维林酮标准品各10mg,分别置于100mL容量瓶中,加溶剂1溶解用溶剂2稀释至刻度,摇匀,作为盐酸屈他维林、屈他维林酮储备液;精密称取乙基罂粟碱标准品20mg,置50mL容量瓶中,加溶剂1溶解用溶剂2稀释至刻度,摇匀,精密量取上述溶液5mL置50mL容量瓶中,加溶剂2稀释至刻度,摇匀,作为乙基罂粟碱储备液。精密量取适量盐酸屈他维林、乙基罂粟碱、屈他维林酮储备液,加溶剂2制成每1mL中含盐酸屈他维林0.16、1.0、2.0、4.0、5.0、6.0μg;乙基罂粟碱0.048、0.8、1.2、1.6、2.0、2.4μg;屈他维林酮0.32、1.0、2.0、4.0、5.0、6.0μg的6份溶液。精密量取上述溶液各20μL分别注入高效液相色谱仪,记录色谱图,测定峰面积,以峰面积A为纵坐标、浓度C为横坐标作线性回归。结果见表5,表5盐酸屈他维林和已知杂质的HPLC线性名称线性方程相关系数线性范围μg/mL盐酸屈他维林A=29.120C+0.5168R2=0.99930.16~6.0乙基罂粟碱A=107.38C+0.8246R2=0.99990.048~2.4屈他维林酮A=23.756C+0.0453R2=0.99960.32~6.0结论:结果表明,盐酸屈他维林在0.16~6.0μg/mL、乙基罂粟碱在0.048~2.4μg/mL、屈他维林酮在0.32~6.0μg/mL范围内,峰面积与测定浓度呈良好线性关系。实施例11供试品溶液稳定性试验取盐酸屈他维林注射液照有关物质检查方法,配制供试品溶液,分别于0、2、4、6、8、10、12h进样,考察乙基罂粟碱、屈他维林酮峰面积、其他最大单杂和杂质总量的变化。结果见表6,表6供试品溶液稳定性试验结果结论:12h内,乙基罂粟碱含量RSD为0.89%,屈他维林酮含量RSD为5.43%,其他最大单杂含量RSD为1.78%,总杂含量RSD为2.51%。从以上测定数据可以看出,供试品溶液在12h内各杂质和总杂无明显变化,稳定性良好。实施例12重复性试验取盐酸屈他维林注射液,照有关物质检查方法,平行配制6份供试品溶液,每份各测定1次,计算各杂质含量。结果见表7,表7重复性试验结果结论:6份供试品溶液测定结果符合要求,重复性良好。实施例13准确度试验分别取盐酸屈他维林注射液1mL,置25mL容量瓶中,共9份,每3份为一组,分别精密加入乙基罂粟碱、屈他维林酮杂质储备液各0.5mL(限度50%)、1.0mL(限度100%)、1.5mL(限度150%),加溶剂2稀释至刻度,作为低、中、高三组浓度供试品溶液,依法测定,计算回收率及RSD。结果见表8和表9,表8乙基罂粟碱准确度试验结果表9屈他维林酮准确度试验结果实施例14耐用性试验有关物质耐用性主要从流动相不同磷酸浓度、不同pH、不同流速和不同色谱柱等几个方面验证。结果见表10,表10耐用性试验结果耐用性试验条件对比图谱见图11-14,结论:综上耐用性试验结果表明,本方法色谱条件参数对盐酸屈他维林注射液有关物质杂质分离与检查没有明显影响。实施例15有关物质检查分别取变更灭菌工艺前、后样品各三批和原研产品,按照国家标准和本发明方法进行有关物质检查,并进行对比研究,综合评估变更灭菌工艺前后样品的杂质变化,新老方法的检测性能。结果见表11,表11灭菌工艺变更前后的盐酸屈他维林注射液与原研产品有关物质杂质比较结论:1、灭菌工艺变更前后各三批样品与原研产品有关物质对比:(1)灭菌工艺变更后的盐酸屈他维林注射液中有1个杂质在原研产品中未出现,含量在0.02%左右;原研产品中有3个杂质在自制样品中未出现;(2)灭菌工艺变更后的盐酸屈他维林注射液中杂质种类和杂质个数均少于原研产品,自制样品和原研产品中均含有乙基罂粟碱和屈他维林酮,自制样品中乙基罂粟碱和屈他维林酮含量均不高于原研产品,自制样品中其他最大单杂含量和杂质总量均不高于原研产品,相同未知杂质均小于原研产品。其他最大单质均小于0.1%。自制品质量不低于原研产品。2、灭菌工艺变更后三批样品和变更前三批样品对比,杂质种类和个数均一致,乙基罂粟碱和屈他维林酮含量无明显变化,其他最大单杂和杂质总量无明显变化。本发明方法和国家局标准(YBH17602006)方法对比如下表12所示:表12本发明方法和国家局标准对比结果采用本发明的原料药盐酸屈他维林有关物质的高效液相色谱分析方法,梯度洗脱,充分揭示了盐酸屈他维林注射液的杂质,提高产品的安全性,方法科学、可靠,操作简便,可控,适于实际应用和推广。最后应说明的是:以上各实施例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述各实施例对本发明进行了详细的说明,本领域的普通技术人员应当理解:其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分或者全部技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的范围。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1