一种远志药材指纹图谱及指标性成分含量测定的方法与流程
本发明属于中药分析及中药材含量测定领域,具体涉及一种远志药材指纹图谱及指标性成分含量测定的方法。
背景技术:
远志polygalatenuifoliawilld.,最早记载于《神农本草经》,列为上品,并被视为养命要药。性温,味苦、辛,具有安神益智、祛痰消肿等功效。经现代研究发现,远志药材的主要有效成分包括皂苷类、糖酯类、口山酮类及生物碱类等,具有抑菌、抗炎、提高认知、改善记忆能力、抗老年痴呆、抗抑郁、抗肿瘤、抗衰老等药理作用。
中药材药效的强弱与药材质量的优劣密切相关,而药材质量的优劣与药材中所含主要化学成分含量的高低紧密相关。中药材中多种已知或未知的化学成分,通过多条途径作用于多靶点发挥其药理作用。目前关于远志药材的定性分析,主要为指纹图谱分析,但之前建立的方法耗时较长,损耗试剂较多,不利于高通量分析,对于复杂成分的指认也不充分;对远志药材中化学成分的定量分析,主要集中于个别成分为指标的测定,单一指标成分难以准确、全面地体现中药材的内在质量,中药多指标质量控制和评价模式需要有各种成分的对照品,但部分中药对照品分离难度大、单体不稳定、难以供应或供应价格高等问题的存在,使其不能满足中药质量监控和新药研发等方面的需求,目前远志药材中已测定含量的化合物数量较少。故针对上述复杂成分指认不充分以及定量化学成分较少等缺陷,本发明提供一种远志药材指纹图谱及指标性成分含量测定的方法,该方法能够较全面地对远志药材中化合物进行定性及定量分析。
技术实现要素:
本发明的目的在于提供一种远志药材指纹图谱及指标性成分含量测定的方法,该方法能够较全面地对远志药材中化合物进行定性及定量分析。
为实现上述目的,本发明提供的技术方案为:
一种远志药材指纹图谱及指标性成分的含量测定方法,步骤如下:
步骤1:采用超高效液相(ultraperformanceliquidchromatography,uplc)全波长可见紫外检测法,建立远志药材的指纹图谱;
混合对照品溶液的制备:分别称取各对照品适量,精密称定,加甲醇溶解,定容至lancerin、sibiricaxanthoneb、glomeratosea、7-o-methylmangiferin、polygalaxanthoneiii终浓度为1-100μg/ml,sibiricosea5、sibiricosea6、tenuifolisidec终浓度为100-300μg/ml,3,6′-disinapoylsucrose、tenuifolisidea终浓度为300-500μg/ml,摇匀,即得混合对照品溶液,并置于4℃冰箱中备用;
供试品溶液的制备:取远志药材粉碎并过药典三号筛,取远志药材粉末约0.2g,精密称定,置具塞锥形瓶中,精密移取70%甲醇溶液20ml,称定重量,超声处理30min(功率500w,频率40khz),放至室温,再称定重量,用70%甲醇溶液补足减失的重量,摇匀,滤过,取续滤液,过0.22μm微孔滤膜,即得;
指纹图谱的测定:分别精密吸取混合对照品溶液和供试品溶液各2-10μl,注入超高效液相色谱仪,记录色谱图,采用国家药典委员会推荐的《中药色谱指纹图谱相似度评价系统》生成远志药材指纹图谱;
其中,色谱条件如下:
色谱柱:uplcc18柱或t3柱;
流动相:流动相a为乙腈,流动相b为体积浓度为0.1-1.0%的甲酸水溶液;
洗脱梯度:0-3min,5-10%a;3-6min,10-12%a;6-16min,12-22%a;16-17min,22%a;17-22min,22-27%a;22-25min,27-32%a;25-27min,32-35%a;27-30min,35%a;30-36min,35-40%a;36-37min,40-100%a;37-39min,100-5%a。
进样量:2-10μl;
柱温:20-40℃;
流速:0.2-0.5ml/min;
检测波长:290-330nm;
步骤2:利用液质联用技术对步骤1中指纹图谱的色谱峰进行指认,确定紫外检测共有的指纹峰及非共有峰,其中液相色谱条件同步骤1,质谱条件如下:
离子源:电喷雾离子源(esi);扫描方式:正负离子同时扫描;喷雾电压:3.5kv;鞘气流速:35psi(1psi≈6.9kpa);辅助气流速:10psi;毛细管温度:320℃;探头加温器温度:300℃;最大喷雾电流:100a;s-lens分辨率:55;扫描范围:m/z100-2000;质量分辨率:70000;
步骤3:从步骤2中指认出的23个共有峰中筛选出8个指标性成分(sibiricosea5、sibiricosea6、sibiricaxanthoneb、glomeratosea、polygalaxanthoneiii、3,6′-disinapoylsucrose、tenuifolisidea、tenuifolisidec),从15个非共有峰中筛选出2个指标性成分(lancerin、7-o-methylmangiferin),采用超高效液相全波长可见紫外检测法对上述10个指标性成分进行含量测定;
测定方法为:分别精密吸取上述混合对照品溶液和供试品溶液各2-10μl,注入超高效液相色谱仪,确定各待测成分的色谱峰,并将混合对照品溶液逐级稀释,建立10个指标性成分的线性回归方程,分别计算供试品溶液中如上所述10个化合物的含量;
其中,色谱条件同步骤1。
进一步,步骤1和3中流动相b为体积浓度为0.1%的甲酸水溶液;进样量:2μl;柱温:40℃;流速:0.4ml/min;检测波长:320nm。
相比于现有技术,本发明通过uplc“指纹图谱定性+指标性成分定量”的方式,从整体和关键组分两方面共同实现质控标准的提升,不仅全面而特定定量的确定了产品的质量可控性,而且由于uplc技术的引入,大大缩短了现有的普遍采用hplc技术的测定时间,使得远志药材指纹图谱的复杂性表征得以实现。
附图说明
图1为本发明提供的7批远志药材指纹图谱及标准指纹图谱;其中样品1-7编号见表1,r为对照指纹图谱;
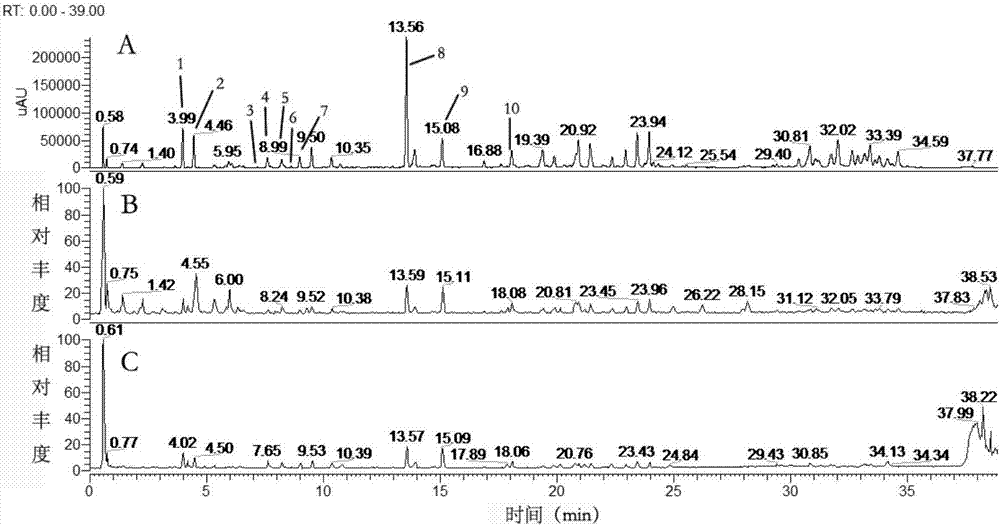
图2为远志样品的液质色谱图;图中:a:紫外色谱图;b:正离子模式下总离子流色谱图;c:负离子模式下总离子流色谱图;
图3为混合对照品溶液的uplc色谱图;
图4为远志药材供试品溶液的uplc色谱图;
图3、图4中:1为sibiricosea5,2为sibiricosea6,3为lancerin,4为sibiricaxanthoneb,5为glomeratosea,6为7-o-methylmangiferin,7为polygalaxanthoneiii,8为3,6′-disinapoylsucrose,9为tenuifolisidea,10为tenuifolisidec)
具体实施方式
以下实施例用于说明本方法,但不用来限制本发明的范围。
为了更好地理解本发明内容,下面结合附图和具体实施例对本发明作进一步说明。
实施例1
步骤1:采用超高效液相全波长可见紫外检测法,建立远志药材的指纹图谱;1.试验材
料、仪器与试剂
试验材料:所用远志药材样本是从山西省不同产地收集到的7批远志药材全根,用手揉搓,抽去木心,得到远志筒,粉碎,过药典三号筛,备用。试验材料具体信息见表1。
表1远志样品信息表
仪器:超高效液相色谱系统(waters公司acquityuplc,包括真空脱气机、二元梯度泵、自动进样器、柱温箱、tuv检测器),电子天平(cpa225d,德国sartorius公司),milli-q超纯水系统(美国millipore公司),超声波清洗器(kq-500dv,昆山市超声仪器有限公司),中药色谱指纹图谱相似度评价系统2008a版。
试剂:乙腈(色谱纯,fisher公司),甲酸(色谱纯,fisher公司),超纯水(milli-q制备),甲醇(国产分析纯)。
2.混合对照品溶液的制备
分别称取各对照品适量,精密称定,加甲醇溶解,定容至lancerin、sibiricaxanthoneb、glomeratosea、7-o-methylmangiferin、polygalaxanthoneiii终浓度为1-100μg/ml,sibiricosea5、sibiricosea6、tenuifolisidec终浓度为100-300μg/ml,3,6′-disinapoylsucrose、tenuifolisidea终浓度为300-500μg/ml的混合对照品溶液,摇匀,即得,并置于4℃冰箱中备用;
3.供试品溶液的制备
取远志药材粉碎并过药典三号筛,取远志药材粉末约0.2g,精密称定,置具塞锥形瓶中,精密移取70%甲醇溶液20ml,称定重量,超声处理30min(功率500w,频率40khz),放至室温,再称定重量,用70%甲醇溶液补足减失的重量,摇匀,滤过,取续滤液,过0.22μm微孔滤膜,即得;
4.指纹图谱的测定
精密吸取供试品溶液2μl,注入超高效液相色谱仪,记录色谱图,即得指纹图谱;
其中,色谱条件如下:
色谱柱:phenomenexkinetexc18(100×2.1mm,2.6μm)色谱柱
流动相:a为乙腈,流动相b为体积浓度为0.1%的甲酸水溶液;
梯度洗脱:0-3min,5-10%a;3-6min,10-12%a;6-16min,12-22%a;16-17min,22%a;17-22min,22-27%a;22-25min,27-32%a;25-27min,32-35%a;27-30min,35%a;30-36min,35-40%a;36-37min,40-100%a;37-39min,100-5%a;
温度:40℃;
流速:0.4ml/min;
色谱图光谱采集范围:190-400nm。
采用“中药色谱指纹图谱相似度评价系统2008a版”软件对7批远志药材uplc指纹图谱进行数据处理,确定共有峰32个(如图1所示),由于3,6′-disinapoylsucrose在各批次远志中均有出现,分离度良好且峰面积较大,故选择3,6′-disinapoylsucrose(9号峰)作为参照峰(s),以该参照峰计算各共有峰相对保留时间和相对峰面积,进行指纹图谱方法学考察,测定方法与步骤1中相同。结果显示,同一样品溶液连续进样6次,各共有峰相对保留时间rsd<0.15%,相对峰面积rsd<0.98%,精密度良好;稳定性考察各共有峰相对保留时间rsd<0.42%,相对峰面积rsd<1.83%;重复性考察各共有峰相对保留时间rsd<0.35%,相对峰面积rsd<2.10%。
7批远志样品的uplc指纹图谱见图1,各产地远志药材指纹图谱与对照指纹图谱比对,相似度介于0.927~0.971,结果见表2。
表27批远志药材间的相似度计算结果(r为对照指纹图谱)
步骤2:利用液质联用技术对步骤1中指纹图谱的色谱峰进行指认,确定紫外检测共有的指纹峰;
1.试验材料、仪器与试剂
试验材料及试剂:同步骤1。
仪器:thermoscientificqexactivelc-ms,美国thermo;其他仪器同步骤1。
2.质谱条件如下:
离子源:电喷雾离子源(esi);扫描方式:正负离子同时扫描;喷雾电压:3.5kv;鞘气流速:35psi(1psi≈6.9kpa);辅助气流速:10psi;毛细管温度:320℃;探头加温器温度:300℃;最大喷雾电流:100a;s-lens分辨率:55;扫描范围:m/z100-2000;质量分辨率:70000;
用步骤1中色谱条件对7批远志药材进行uplc指纹图谱分析。对各样品的32个共有峰紫外光谱图进行对比分析,所得紫外图谱如图1所示。
采用国家药典委员会推荐的《中药色谱指纹图谱相似度评价系统》生成远志药材标准指纹图谱,其中有32个共有峰,峰号依次记为1-32号。在指纹图谱中,以3,6′-disinapoylsucrose(9号峰)为参照峰s峰,其中1、2、3、4、5、9、11、14号峰采用对照品色谱及质谱保留行为进行指认,依次为sibiricosea5、sibiricosea6、sibiricaxanthoneb、glomeratosea、polygalaxanthoneiii、3,6′-disinapoylsucrose、tenuifolisidea、tenuifolisidec,由于化合物lancerin及7-o-methylmangiferin的含量较低,在指纹图谱中没有这两个化合物的共有峰,可通过对照品色谱及质谱保留行为,并依据参考文献对两个化合物进行指认。
从远志药材总离子流图可以看出(图2),其各成分峰在正、负离子条件下均有较高的响应,并与紫外色谱图对应。
依据对照品色谱及质谱保留行为,并参考文献中的数据和色谱保留行为(色谱图见图2),对32个共有峰中的23个进行了鉴定(见表3),包括14个糖酯类成分,6个皂苷类成分,以及3个口山酮类成分。同时参考文献中的数据和色谱保留行为对远志样品中其他15个非共有峰进行了归属,包括9个糖酯类成分以及6个口山酮类成分,结果见表4,。
表3远志指纹图谱共有峰的归属(负离子模式)
表4基于液质指认的远志指纹图谱中非共有峰的归属(负离子模式)
步骤3:从步骤2中指认的23个共有峰中筛选出8个指标性成分(sibiricosea5、sibiricosea6、sibiricaxanthoneb、glomeratosea、polygalaxanthoneiii、3,6′-disinapoylsucrose、tenuifolisidea、tenuifolisidec),从15个非共有峰中筛选出2个指标性成分(lancerin、7-o-methylmangiferin),采用超高效液相全波长可见紫外检测法对上述10个指标性成分进行含量测定。
1.试验材料、仪器与试剂同步骤1。
2.混合对照品溶液及供试品溶液的制备同步骤1。
3.指标性成分的含量测定
分别精密吸取上述混合对照品溶液和供试品溶液各2μl,注入超高效液相色谱仪,按步骤1中色谱条件分别对混合对照品溶液和供试品溶液进行测定,确定各待测成分的色谱峰,并将混合对照品溶液逐级稀释,建立10个指标性成分的线性回归方程,分别计算供试品溶液中sibiricosea5、sibiricosea6、lancerin、sibiricaxanthoneb、glomeratosea、7-o-methylmangiferin、polygalaxanthoneiii、3,6′-disinapoylsucrose、tenuifolisidea、tenuifolisidec10个指标性成分的含量,其中,10个指标性成分的线性回归方程见表5。
表5远志药材中10种指标性成分的线性回归方程及线性范围
其中,x为相应成分在相应检测波长处检测得到的吸光度
4.精密度试验精密量取步骤1中混合对照品溶液2μl,按步骤1中的色谱条件进行测定,连续进样6次,记录上述10个化合物的色谱峰面积,并计算10个化合物保留时间及峰面积rsd值,保留时间的rsd值分别为0.20%、0.26%、0.30%、0.33%、0.35%、0.32%、0.34%、0.29%、0.26%、0.33%;峰面积的rsd值分别为0.41%、0.59%、0.84%、0.61%、0.87%、0.80%、0.38%、0.70%、0.91%、0.55%,表明仪器精密度良好。
5.稳定性试验取同一批样品(3号),按步骤1中备样方法平行制备6份,室温下放置,按步骤1中色谱条件分别在0、2、4、8、12、18、24h时进样分析,计算10个化合物保留时间及峰面积的rsd值,保留时间的rsd值分别为0.62%、0.60%、0.52%、0.43%、0.42%、0.41%、0.36%、0.21%、0.17%、0.16%;峰面积的rsd值分别为0.86%、0.95%、1.63%、0.60%、1.50%、0.48%、0.48%、0.45%、0.84%、0.34%,表明样品在24h内稳定性良好。
6.重复性试验取同一批样品(3号),按步骤1中备样方法平行制备6份,按步骤1中色谱条件进样分析,计算10个化合物保留时间及质量分数rsd值,保留时间的rsd值分别为0.57%、0.56%、0.47%、0.55%、0.49%、0.50%、0.46%、0.30%、0.28%、0.33%;质量分数的rsd值分别为1.43%、1.22%、2.00%、0.70%、1.99%、1.67%、2.99%、1.08%、0.88%、1.22%,表明实验重复性良好。
7.加样回收率试验取已测定的3号样品6份,每份为0.1g,分成3组,每组3份,精密称定,置于具塞锥形瓶中,分别精密加入相当于样品成分量50%、100%、150%的各对照品溶液,按步骤1中备样方法制备供试品溶液,按步骤1中色谱条件进样分析,计算10种成分的加样回收率,平均回收率分别为98.45%、99.17%、95.76%、95.72%、95.44%、92.29%、91.85%、99.18%、96.11%、94.91%;rsd分别为0.96%、1.78%、1.68%、1.77%、1.72%、1.70%、0.82%、1.38%、1.16%、1.82%,符合2015版《中国药典》中《药品质量标准分析方法验证指导原则》的要求。
8.样品含量测定按步骤1中备样方法制备7批供试品溶液,按步骤1中色谱条件分别测定7批远志样品的10种成分的含量,并计算每批样品10种成分的总含量。7批远志药材中10个指标性成分含量测定结果见表6,混合对照品的色谱图如图3所示,远志样品的色谱图如图4所示。
表67批远志中10种化学成分的含量
虽然,上文中已经用一般性说明及具体实施方案对本发明作了详尽的描述,但在本发明基础上,可以对之作一些修改或改进,这对本领域技术人员而言是显而易见的。因此,在不偏离本发明精神的基础上所做的这些修改或改进,均属于本发明要求保护的范围。
- 还没有人留言评论。精彩留言会获得点赞!