一种检测内毒素的方法与流程
本发明涉及一种检测内毒素的方法。
背景技术:
内毒素(lps)是一种高毒性的炎症刺激物,它可以与特异性细胞受体发生相互作用,从而产生炎性细胞因子,会导致发烧、败血症、多器官衰竭甚至死亡。由于可能产生严重的免疫反应,因此检测lps变得至关重要,尤其是注射液等生物制品等,以便于确保灭菌产品的安全。目前用于lps检测的主要方法是鲎试剂法(lal),在许多领域中都被广泛使用。[2]近年来还有一些基于lps抗体或适配体的电化学和光学传感器的研究,[3,4]它们能够特异性地鉴别lps,并且克服一些传统方法的缺点。适配体是具有高特异性和与靶标结合的高亲和性的单链寡核苷酸(ssrna或ssdna),它可以通过折叠成独特的三维结构从而与靶分子结合,范围从小分子到蛋白质甚至细胞。[5]最近也已经有较多研究筛选出lps的适配体。
虽然鲎试剂法是目前测定lps的金标准,但是它对温度、ph的变化以及干扰因子都非常敏感,并且样品制备程序繁琐。更重要的是,该方法依赖于因过度捕捞而严重减少的动物鲎。而基于lps抗体、适配体的生物传感器虽然能够特异性识别lps,但是其实际应用仍然受到成本高,耗时长,且灵敏度低的限制。迄今为止,lps的检测灵敏度虽然已从毫摩尔水平提高到纳摩尔水平,但在飞摩尔水平检测lps仍然是一个挑战。对于石墨烯,如果仅通过石墨烯来制造传感器,则不能很好地控制反应条件。例如,石墨烯的生物相容性非常低,这会影响其生物识别过程。最重要的是,石墨烯传感器的选择性和灵敏度都会受到限制。因此,研究一种检测lps的新方法非常迫切。
技术实现要素:
这了解决上述存在问题,本发明通过研究,基于内毒素适配体构建了一种检测lps的生物传感器,具体开发了一种基于pdms芯片进行连续进样检测lps的方法。该检测系统将微流控芯片平台与适配体相结合,能够检测各种实际样品中的lps,且所需时间更短(1h),实验设置更简单。
本发明的目的在于提供一种检测内毒素的方法。
本发明所采取的技术方案是:
一种检测内毒素的方法,包括以下步骤:
1)将待测样品与荧光标记的lps适配体,进行第一次混合孵育;再加入还原氧化石墨烯和tbe缓冲液,进行第二次混合孵育;
2)将微流控芯片清洗干净后,用含ch3oh的tbe缓冲液充满整个微流控芯片的通道,然后将上步第二次混合孵育的样品加样到微流控芯片的进样口中,在微流控芯片的进、出样口之间施加电压进行处理,进样口处为阳极,电压处理后检测nafion膜通道与阳极之间的荧光强度;根据荧光强度和标准曲线判定待测样品中lps的含量;
上述微流控芯片中的nafion膜通道垂直于微流控芯片中的富集通道。
进一步的,所述待测样品包括血清样品、水溶液样品。
进一步的,步骤1)中,当待测样品为血清样品时,在待测样品与lps适配体混合之前,还需加入sds、β-巯基乙醇,并于92~100℃加热处理3~8min。
进一步的,所述sds、β-巯基乙醇在待测样品中的浓度分别为7~10mg/ml、3~10%v/v。
更进一步的,sds浓度为10mg/ml,β-巯基乙醇浓度为5%v/v。
进一步的,步骤1)中,所述荧光标记的lps适配体在加入待测样品之前,于92~100℃加热5~13min,并再于10~20℃冷却5~12min。更进一步的,lps适配体加热时间为5~10min,最佳的加热时间为10min;最佳的冷却时间为10min。
进一步的,步骤1)中,第一次混合孵育的温度为10~20℃,孵育时间为7~14min。
进一步的,步骤1)中,第二次混合孵育的时间不少于25min,温度为室温;第二次混合孵育时间在30min以上,检测效果会更佳。
进一步的,步骤1)中,第二次混合孵育体系中,还原氧化石墨烯的浓度为8~17μg/ml、荧光标记的lps适配体浓度为4~8nm。
进一步的,步骤2)中,所述tbe缓冲液的ph为7~7.8,tbe缓冲液中含有20~30%v/vch3oh。
进一步的,步骤2)中,当待测样品为血清样品时,所述tbe缓冲液ph为7~7.8,含有20~30%v/vch3oh和4~10%v/vch3cn。更进一步的,ch3cn的浓度为5%v/v时,检测效果会更佳。
进一步的,步骤2)中,电压处理时间为25~35min,电压为25~35v。
进一步的,步骤2)中,nafion膜通道的宽度为200~400μm,深度为20~200μm。
更进一步的,nafion膜通道的宽度为400μm,nafion膜通道的深度为45μm时,检测效果会更佳。
本发明的有益效果是:
(1)本发明建立了一种检测样品中内毒素的新方法,该方法的最低检测限(lod)为8fm,线性范围为:50fm-1nm(水溶液样品),50fm-50pm(血浆样品)。本发明灵敏度高,选择性好,抗干扰能力较强。
(2)本发明方法检测所需时间短,消耗试剂量少,且不依赖于动物来源
(3)本发明能够检测注射液及脓毒症模型小鼠血清中的lps含量,能快速区分革兰氏阳性菌、革兰氏阴性菌、真菌。
附图说明
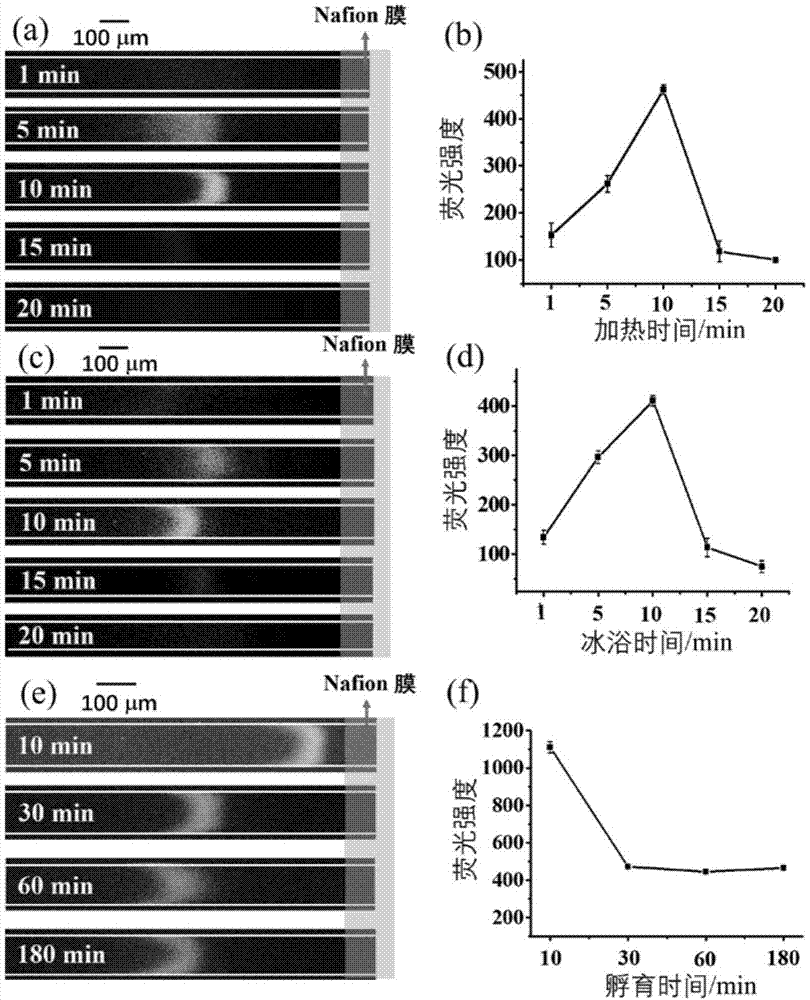
图1.lps适配体的加热时间、冰浴时间以及与rgo孵育时间的对本发明检测lps的影响;(a)适配体加热时间对检测荧光条带的影响图;(b)适配体加热时间对检测荧光强度影响的曲线图;(c)lps适配体冰浴时间对检测荧光条带的影响图;(d)lps适配体冰浴时间对检测荧光强度影响的曲线图;(e)rgo的孵育时间对检测荧光条带的影响图;(f)rgo的孵育时间对检测荧光强度影响的曲线图。
图2.在teb缓冲液中使用有机溶剂ch3cn对本发明检测lps效果的影响。(a)ch3cn的浓度对检测荧光条带的影响图;(b)ch3cn浓度对检测荧光强度影响的曲线图。
图3.nafion膜尺寸对对本发明检测lps效果的影响。(a)nafion膜通道的宽度对检测荧光条带的影响图;(b)nafion膜通道的宽度对检测荧光强度影响的曲线图;(c)nafion膜通道的深度对检测荧光条带的影响图;(d)nafion膜通道的深度对检测荧光强度影响的曲线图。
图4.sds和β-巯基乙醇对检测lps效果的影响;(a)(b)为sds浓度对富集荧光条带的影响:(a)为样品,(b)为空白(无lps);(c)(d)为β-巯基乙醇对富集荧光强度的影响:(c)样品,(d)为空白(无lps)。
图5.本发明方法检测对脓毒症模型小鼠血清中lps的检测。
图6.本发明方法的特异性检测;(a)和(b)分别为lps水溶液样品和lps血清样品对该方法的选择性。红色柱表示样品中同时存在竞争分子与lps,黑色柱表示样品中只存在竞争分子。(c)30v电压下三种菌液(108cfu/ml)在ci-es平台的富集荧光强度。
图7.本发明方法检测lps的荧光强度(y)与相应的浓度(x)的关系的标准曲线。(a)为水溶液样品,(b)为血清样品。
具体实施方式
下面结合具体实施例对本发明作进一步的说明。
实施例1一种检测内毒素的方法
1)微流控芯片的制作:本实施例采用pdms(聚二甲基硅氧烷)微流控芯片,其中含有nafion膜,nafion膜垂直于微流控芯片中的通道。微流控芯片的具体制作过程如下:
在真空干燥器中用三甲基氯硅烷处理硅片,以避免pdms粘附到硅片模具上。pdms与固化剂以重量比为10:1的比例充分混合均匀,放入真空干燥器中真空脱气至气泡完全消失后,缓慢浇注在硅片上,避免产生气泡。然后在95℃的加热板上固化2-3h,然后将pdms从硅片上剥离,这样硅片上的微结构就转移到了具有弹性的pdms上,并在进样口和出样口处打孔。之后在1mnaoh亲水处理过的载玻片上制作nafion膜:采用45μm厚,400μm宽的pdms微通道,通道两端各有一个孔,将1μlnafion膜液加到直通道的入口中,并在直通道出口处用注射器吸使nafion膜液充满整个直通道。放置3min后迅速剥离pdms通道,将含有nafion膜的载玻片在95℃下加热5min后使其固化,然后将含有通道的pdms芯片与含有nafion膜的载玻片一起放入等离子清洗机中进行表面改性处理,最后将pdms芯片不可逆地粘合到载玻片上,nafion膜垂直于pdms芯片的富集通道。
2)样品处理:首先将6-fam(6-羧基荧光素)标记的lps适配体在95℃沸水中加热10min,然后立即放入15℃水中冷却10min。而后将其与待测样品(如果待测样品为血清样品时,在待测样品与lps适配体混合之前,还需将待测样品中加入sds7~10mg/ml、β-巯基乙醇3~10%v/v,并于95℃加热处理5min)混合并继续在15℃进行第一次孵育10min。然后再加入还原氧化石墨烯rgo和1×tbe缓冲液,进行第二次混合孵育,即在室温下旋转孵育30min后上样进行检测。
在上述第二次混合孵育体系中,还原氧化石墨烯的浓度为8~17μg/ml、荧光标记的lps适配体浓度为4~8nm。
上述rgo采用水热还原法制备:将50ml0.05mg/mlgo(氧化石墨烯)水溶液加入到聚四氟乙烯衬里的高压釜中,然后在180℃下加热6h。然后将高压釜冷却至室温。
3)微流控芯片检测:实验之前,用1%bsa(牛血清白蛋白)在室温下修饰微通道10min,以防止非特异性粘附,然后将通道用超纯水洗涤三次并用tbe缓冲液(ph为7~7.8,含有20~30%v/vch3oh;如果待测样品为血清样品时,1×tbe缓冲液含有20~30%v/vch3oh和4~10%v/vch3cn)充满整个通道直至开始实验。将1-10μl枪头切去尖端后作为样品池插入进、出样口中。将上步第二次混合孵育的样品加样到微流控芯片的样品池中,微流控芯片中含有nafion膜(厚45μm,宽400μm);同时将连接dc电源的铂电极插入样品池中,在微流控芯片的进、出样口之间施加20~30v电压进行处理,进样口处为阳极,电压处理30min后检测nafion膜与阳极之间的荧光强度;根据荧光强度和标准曲线判定待测样品中lps的含量。
上述荧光强度的具体测量过程为:通过imagej软件分析荧光图像,使用imagej的subtract函数从图像中扣除背景空白值。然后测量nafion膜与阳极之间的浓度荧光条带(image→adjust→threshold)以得到荧光强度。
实施例2一种检测内毒素的方法(lps适配体的加热时间、冰浴时间以及与rgo孵育时间的优化)
1)微流控芯片的制作:同实施例1
2)样品处理:
lps标准溶液的制备:用超纯水制备lps的标准储备液(1mg/ml)。通过适当稀释储备溶液以制备不同浓度的lps溶液。本实施例以10pm的lps溶液作为后续待测样品。
取6-fam(6-羧基荧光素)标记的lps适配体(由生物公司设计合成)在95℃水浴分别加热1、5、10、12、20min,然后立即放入15℃水中分别冰浴1、5、10、12、20min。而后将其与待测样品(10pm的lps溶液,lps终浓度为1pm)混合并继续在15℃进行第一次孵育10min。然后再加入还原氧化石墨烯rgo和1×tbe缓冲液(ph7.4),进行第二次混合孵育,即在室温下分别旋转孵育10、30、60、180min后上样进行检测。
在上述第二次混合孵育体系中,还原氧化石墨烯的浓度为10μg/ml、荧光标记的lps适配体浓度为6nm。
3)微流控芯片检测:实验之前,用1%bsa(牛血清白蛋白)在室温下修饰微通道10min,以防止非特异性粘附,然后将通道用超纯水洗涤三次并用tbe缓冲液(ph7.4,含有25%v/vch3oh)充满整个通道直至开始实验。将1-10μl枪头切去尖端后作为样品池插入进、出样口中。将上步第二次混合孵育的样品加样到微流控芯片的样品池中,微流控芯片中含有nafion膜通道(nafion膜通道尺寸:深45μm,宽400μm);同时将连接dc电源的铂电极插入样品池中,在微流控芯片的进、出样口之间施加30v电压进行处理,进样口处为阳极,电压处理30min后检测各组中nafion膜与阳极之间的荧光强度。
上述荧光强度的具体测量过程为:通过imagej软件分析荧光图像,使用imagej的subtract函数从图像中扣除背景空白值。然后测量nafion膜与阳极之间的浓度荧光条带(image→adjust→threshold)以得到荧光强度。
本实施例分别探究了lps适配体的加热时间、冰浴时间以及与rgo孵育时间的最佳条件,检测结果如图1所示,从中可以看出,图(a)和(b)表明lps适配体加热时间为5~10min、10~13min时,能够较好检测出样品中的lps,其中最佳的加热时间为10min;图(c)和(d)表明lps适配体冰浴时间为5~12min时,能够较好检测出样品中的lps,其中最佳的冰浴时间为10min;图(e)和(f)表明与rgo孵育时间为25min以上,能够较好检测出样品中的lps,其中孵育时间在30min以上,检测效果会更佳。
实施例3一种检测内毒素的方法(ch3cn的效果检测)
1)微流控芯片的制作:同实施例1
2)样品处理:
lps加标血清的制备:采集健康人的血清,加入实施例2中已知浓度的lps标准储备液,将混合物在95℃沸水中加热5min,即得lps加标血清,将该加标血清加入sds10mg/ml,β-巯基乙醇5%v/v,并于95℃沸水中加热5min,作为后续的待检测样品。
取6-fam(6-羧基荧光素)标记的lps适配体(由生物公司设计合成)在95℃水浴加热10min,然后立即放入15℃水中分别冰浴10min。而后将其与上述待测样品混合并继续在15℃进行第一次孵育10min。然后再加入还原氧化石墨烯rgo以及1×tbe缓冲液,进行第二次混合孵育,即在室温下旋转孵育30min后上样进行检测。
在上述第二次混合孵育体系中,还原氧化石墨烯的浓度为10μg/ml、荧光标记的lps适配体浓度为6nm。
3)微流控芯片检测:实验之前,用1%bsa(牛血清白蛋白)在室温下修饰微通道10min,以防止非特异性粘附,然后将通道用超纯水洗涤三次并用不同的1×tbe缓冲液(ph7.4,含有25%v/vch3oh,ch3cn的浓度分别设置为0%、1%、3%、5%、7%、10%v/v)充满整个通道直至开始实验。将1-10μl枪头切去尖端后作为样品池插入进、出样口中。将上步第二次混合孵育的样品加样到微流控芯片的样品池中,微流控芯片中含有nafion膜(厚45μm,宽400μm);同时将连接dc电源的铂电极插入样品池中,在微流控芯片的进、出样口之间施加20v电压进行处理,进样口处为阳极,电压处理30min后检测各组中nafion膜与阳极之间的荧光强度。
上述荧光强度的具体测量过程为:通过imagej软件分析荧光图像,使用imagej的subtract函数从图像中扣除背景空白值。然后测量nafion膜与阳极之间的浓度荧光条带(image→adjust→threshold)以得到荧光强度。
本实施例分别探究了ch3cn本发明检测lps效果的影响,检测结果如图2所示,从中可以看出,图(a)和(b)表明血清样品基质比水溶液样品更复杂,我们发现在tbe缓冲液中添加ch3cn可改善带状稳定性,当没有ch3cn时,荧光条带较为松散。当ch3cn的浓度为4~10%v/vch3cn时,检测效果较好,当ch3cn的浓度为5%v/v时,检测效果会更佳。
实施例4一种检测内毒素的方法(nafion膜通道的深度和宽度的优化)
1)微流控芯片的制作:除了将nafion通道的尺寸宽度分别设置成100、200、400、600、800μm,深度设置成20、45、100、200μm;其他均同实施例1
2)样品处理:
lps标准溶液的制备:用超纯水制备lps的标准储备液(1mg/ml)。通过适当稀释储备溶液以制备不同浓度的lps溶液。本实施例以10pm的lps溶液作为后续待测样品。
取6-fam(6-羧基荧光素)标记的lps适配体(由生物公司设计合成)在95℃水浴加热10min,然后立即放入15℃水中分别冰浴10min。而后将其与上述待测样品(10pm的lps溶液)混合并继续在15℃进行第一次孵育10min。然后再加入还原氧化石墨烯rgo及1×tbe缓冲液(ph7.4),进行第二次混合孵育,即在室温下旋转孵育30min后上样进行检测。
在上述第二次混合孵育体系中,还原氧化石墨烯的浓度为10μg/ml、荧光标记的lps适配体浓度为6nm。
3)微流控芯片检测:实验之前,用1%bsa(牛血清白蛋白)在室温下修饰微通道10min,以防止非特异性粘附,然后将通道用超纯水洗涤三次并用tbe缓冲液(含25%v/vch3oh)充满整个通道直至开始实验。将1-10μl枪头切去尖端后作为样品池插入进、出样口中。将上步第二次混合孵育的样品加样到微流控芯片的样品池中,微流控芯片中含有nafion膜;同时将连接dc电源的铂电极插入样品池中,在微流控芯片的进、出样口之间施加30v电压进行处理,进样口处为阳极,电压处理30min后检测各组中nafion膜与阳极之间的荧光强度。
上述荧光强度的具体测量过程为:通过imagej软件分析荧光图像,使用imagej的subtract函数从图像中扣除背景空白值。然后测量nafion膜与阳极之间的浓度荧光条带(image→adjust→threshold)以得到荧光强度。
本实施例分别探究了nafion膜通道的宽度和深度对检测的荧光条带和荧光强度的影响,检测结果如图3所示,从中可以看出,图(a)和(b)表明nafion膜通道的宽度为200~400μm时,检测到的荧光条带较清晰、集中,检测到的荧光强度较高,其中最佳nafion膜通道的宽度为400μm;图(c)和(d)表明nafion膜通道的深度为20~200μm时,检测到的荧光条带较清晰、集中,检测到的荧光强度较高,其中最佳nafion膜通道的深度为45μm。
实施例5一种检测内毒素的方法(β-巯基乙醇和sds的效果检测)
1)微流控芯片的制作:同实施例1
2)样品处理:
lps加标血清的制备:采集健康人体的血清,加入实施例2中已知浓度的lps标准储备液,即得lps加标血清,然后将上述加标血清中分别加入sds的浓度为7、10、13、18mg/ml;以及分别加入β-巯基乙醇的浓度为3、5、7、10、15%v/v,将各组混合物在95℃沸水中加热5min,分别作为后续的待检测样品。
取6-fam(6-羧基荧光素)标记的lps适配体(由生物公司设计合成)在95℃水浴加热10min,然后立即放入15℃水中分别冰浴10min。而后将其与上述各组待测样品混合并继续在15℃进行第一次孵育10min。然后再加入还原氧化石墨烯rgo以及1×tbe缓冲液(ph7.4),进行第二次混合孵育,即在室温下旋转孵育30min后上样进行检测。
在上述第二次混合孵育体系中,还原氧化石墨烯的浓度为10μg/ml、荧光标记的lps适配体浓度为6nm。
3)微流控芯片检测:实验之前,用1%bsa(牛血清白蛋白)在室温下修饰微通道10min,以防止非特异性粘附,然后将通道用超纯水洗涤三次并用1×tbe缓冲液(ph7.4,含有25%v/vch3oh和5%v/vch3cn)充满整个通道直至开始实验。将1-10μl枪头切去尖端后作为样品池插入进、出样口中。将上步第二次混合孵育的样品加样到微流控芯片的样品池中,微流控芯片中含有nafion膜(厚45μm,宽400μm);同时将连接dc电源的铂电极插入样品池中,在微流控芯片的进、出样口之间施加20v电压进行处理,进样口处为阳极,电压处理30min后检测各组中nafion膜与阳极之间的荧光强度。
上述荧光强度的具体测量过程为:通过imagej软件分析荧光图像,使用imagej的subtract函数从图像中扣除背景空白值。然后测量nafion膜与阳极之间的浓度荧光条带(image→adjust→threshold)以得到荧光强度。
本实施例探究了sds和β-巯基乙醇对检测lps效果的影响,检测结果如图4所示,从中可以看出图(a)和(b)表明sds浓度为7~10mg/ml时,检测到的荧光条带清晰、集中,最佳的sds浓度为10mg/ml,且空白组基本无荧光富集。图(c)和(d)表明β-巯基乙醇浓度为3~10%v/v时,检测到的荧光条带清晰、集中,最佳β-巯基乙醇浓度为5%v/v,且空白组基本无荧光富集;上述结果说明通过加入sds和β-巯基乙醇能够降低血浆样品荧光的干扰,且整个检测过程只需要5min。
实施例6一种检测内毒素的方法(动物实验)
1)微流控芯片的制作:同实施例1
2)样品处理:
lps血清样品的制备(动物实验):取南方医科大学实验动物中心6-7周无特定病原体(spf)km小鼠。实验时它们的重量为31.6±1.0g。所有实验均按照国家实验室动物护理和使用指南进行。在动物实验中,腹腔注射15mg/kg的lps,然后分别在注射后4h和12h时取0.3ml血液用于lps试验。经测量,注射lps后小鼠肛温由36.2℃降至32.2℃。将获得的血液样品以300g离心10min,分离获得lps血清样品,之后将上述小鼠血清加入10mg/mlsds,5%v/vβ-巯基乙醇,并于95℃沸水中加热5min。作为后续的待测样品。
取6-fam(6-羧基荧光素)标记的lps适配体(由生物公司设计合成)在95℃水浴加热10min,然后立即放入15℃水中分别冰浴10min。而后将其与上述待测样品混合并继续在15℃进行第一次孵育10min。然后再加入还原氧化石墨烯rgo以及1×tbe缓冲液(ph7.4),进行第二次混合孵育,即在室温下旋转孵育30min后上样进行检测。
在上述第二次混合孵育体系中,还原氧化石墨烯的浓度为10μg/ml、荧光标记的lps适配体浓度为6nm。
3)微流控芯片检测:实验之前,用1%bsa(牛血清白蛋白)在室温下修饰微通道10min,以防止非特异性粘附,然后将通道用超纯水洗涤三次并用tbe缓冲液(ph7.4,含有25%v/vch3oh和5%v/vch3cn)充满整个通道直至开始实验。将1-10μl枪头切去尖端后作为样品池插入进、出样口中。将上步第二次混合孵育的样品加样到微流控芯片的样品池中,微流控芯片中含有nafion膜(厚45μm,宽400μm);同时将连接dc电源的铂电极插入样品池中,在微流控芯片的进、出样口之间施加20v电压进行处理,进样口处为阳极,电压处理30min后检测各组中nafion膜与阳极之间的荧光强度。
上述荧光强度的具体测量过程为:通过imagej软件分析荧光图像,使用imagej的subtract函数从图像中扣除背景空白值。然后测量nafion膜与阳极之间的浓度荧光条带(image→adjust→threshold)以得到荧光强度。
检测结果如图5所示,可见利用本发明方法分别对注射lps4h及12h后的脓毒症模型小鼠血清进行测定,均能检测出小鼠血清中的lps,且对照组基本无荧光富集。
实施例7一种检测内毒素的方法(细菌实验及特异性检测)
方法:
1)微流控芯片的制作:同实施例1
2)样品处理:
①基体为水溶液:
空白组:已知浓度的lps标准水溶液,作为待测样品。
取6-fam(6-羧基荧光素)标记的lps适配体(由生物公司设计合成)在95℃水浴加热10min,然后立即放入15℃水中冰浴10min。而后将其与上述待测样品混合并继续在15℃进行第一次孵育10min。然后再加入还原氧化石墨烯rgo以及1×tbe缓冲液(ph7.4),进行第二次混合孵育,即在室温下旋转孵育30min后上样进行检测。
焦磷酸盐组:已知浓度的lps标准水溶液+同浓度焦磷酸盐,作为待测样品。
取6-fam(6-羧基荧光素)标记的lps适配体(由生物公司设计合成)在95℃水浴加热10min,然后立即放入15℃水中冰浴10min。而后将其与上述待测样品混合并继续在15℃进行第一次孵育10min。然后再加入还原氧化石墨烯rgo以及1×tbe缓冲液(ph7.4),进行第二次混合孵育,即在室温下旋转孵育30min后上样进行检测。
fad+组:已知浓度的lps标准水溶液+同浓度fad+,作为待测样品。
取6-fam(6-羧基荧光素)标记的lps适配体(由生物公司设计合成)在95℃水浴加热10min,然后立即放入15℃水中冰浴10min。而后将其与上述待测样品混合并继续在15℃进行第一次孵育10min。然后再加入还原氧化石墨烯rgo以及1×tbe缓冲液(ph7.4),进行第二次混合孵育,即在室温下旋转孵育30min后上样进行检测。
nad+组:已知浓度的lps标准水溶液+同浓度nad+,作为待测样品。
取6-fam(6-羧基荧光素)标记的lps适配体(由生物公司设计合成)在95℃水浴加热10min,然后立即放入15℃水中冰浴10min。而后将其与上述待测样品混合并继续在15℃进行第一次孵育10min。然后再加入还原氧化石墨烯rgo以及1×tbe缓冲液(ph7.4),进行第二次混合孵育,即在室温下旋转孵育30min后上样进行检测。
amp组:已知浓度的lps标准水溶液+同浓度amp,作为待测样品。
取6-fam(6-羧基荧光素)标记的lps适配体(由生物公司设计合成)在95℃水浴加热10min,然后立即放入15℃水中冰浴10min。而后将其与上述待测样品混合并继续在15℃进行第一次孵育10min。然后再加入还原氧化石墨烯rgo以及1×tbe缓冲液(ph7.4),进行第二次混合孵育,即在室温下旋转孵育30min后上样进行检测。
adp组:已知浓度的lps标准水溶液+同浓度adp,作为待测样品。
取6-fam(6-羧基荧光素)标记的lps适配体(由生物公司设计合成)在95℃水浴加热10min,然后立即放入15℃水中冰浴10min。而后将其与上述待测样品混合并继续在15℃进行第一次孵育10min。然后再加入还原氧化石墨烯rgo以及1×tbe缓冲液(ph7.4),进行第二次混合孵育,即在室温下旋转孵育30min后上样进行检测。
atp组:已知浓度的lps标准水溶液+同浓度atp,作为待测样品。
取6-fam(6-羧基荧光素)标记的lps适配体(由生物公司设计合成)在95℃水浴加热10min,然后立即放入15℃水中冰浴10min。而后将其与上述待测样品混合并继续在15℃进行第一次孵育10min。然后再加入还原氧化石墨烯rgo以及1×tbe缓冲液(ph7.4),进行第二次混合孵育,即在室温下旋转孵育30min后上样进行检测。
磷脂酰胆碱组:已知浓度的lps标准水溶液+同浓度磷脂酰胆碱,作为待测样品。
取6-fam(6-羧基荧光素)标记的lps适配体(由生物公司设计合成)在95℃水浴加热10min,然后立即放入15℃水中冰浴10min。而后将其与上述待测样品混合并继续在15℃进行第一次孵育10min。然后再加入还原氧化石墨烯rgo以及1×tbe缓冲液(ph7.4),进行第二次混合孵育,即在室温下旋转孵育30min后上样进行检测。
脂磷壁酸组:已知浓度的lps标准水溶液+同浓度脂磷壁酸,作为待测样品。
取6-fam(6-羧基荧光素)标记的lps适配体(由生物公司设计合成)在95℃水浴加热10min,然后立即放入15℃水中冰浴10min。而后将其与上述待测样品混合并继续在15℃进行第一次孵育10min。然后再加入还原氧化石墨烯rgo以及1×tbe缓冲液(ph7.4),进行第二次混合孵育,即在室温下旋转孵育30min后上样进行检测。
β-d-葡聚糖组:已知浓度的lps标准水溶液+同浓度β-d-葡聚糖,作为待测样品。
取6-fam(6-羧基荧光素)标记的lps适配体(由生物公司设计合成)在95℃水浴加热10min,然后立即放入15℃水中冰浴10min。而后将其与上述待测样品混合并继续在15℃进行第一次孵育10min。然后再加入还原氧化石墨烯rgo以及1×tbe缓冲液(ph7.4),进行第二次混合孵育,即在室温下旋转孵育30min后上样进行检测。
②基体为血清:除了需要在lps加标血清中加入10mg/mlsds和5%v/vβ-巯基乙醇在沸水中进行加热处理5min与①不同,其他均与①相同。
③大肠杆组:通过4,000rpm离心10min收集大肠杆菌,并用1×tbe(ph7.4)洗涤3遍,然后以13,000rpm离心10min破碎细菌,取上清后稀释于1×tbe溶液中(c=108cfu/ml)作为待测样品。
④金黄色葡萄球菌组:通过4,000rpm离心10min收集金黄色葡萄球菌,并用1×tbe(ph7.4)洗涤3遍,然后以13,000rpm离心10min破碎细菌,取上清后稀释于1×tbe溶液中(c=108cfu/ml)作为待测样品。
⑤白色念珠菌组:通过4,000rpm离心10min收集白色念球菌,并用1×tbe(ph7.4)洗涤3遍,然后以13,000rpm离心10min破碎细菌,取上清后稀释于1×tbe溶液中(c=108cfu/ml)作为待测样品。
取6-fam(6-羧基荧光素)标记的lps适配体(由生物公司设计合成)在95℃水浴加热10min,然后立即放入15℃水中分别冰浴10min。而后将其与上述各待测样品混合并继续在15℃进行第一次孵育10min。然后再加入还原氧化石墨烯rgo以及1×tbe缓冲液(ph7.4,),进行第二次混合孵育,即在室温下旋转孵育30min后上样进行检测。
在上述第二次混合孵育体系中,还原氧化石墨烯的浓度为10μg/ml、荧光标记的lps适配体浓度为6nm。
3)微流控芯片检测:同实施例2。
检测结果如图6所示,从中可以看出,本发明方法具有较好的特异性。在一些竞争生物分子(焦磷酸盐,fad+,nad+,amp,adp,atp,磷脂酰胆碱,lta,β-d-葡聚糖)的存在下,根据其荧光强度来评估其选择性,如图6(a)和(b)所示,所有竞争性生物分子均不会引起荧光强度的显著增加。我们所用的是已报道的能够特异性识别lps的适配体,因此,只有lps能诱导荧光强度发生显著的变化。该方法还能够区分革兰氏阴性菌、革兰氏阳性菌和真菌。我们使用大肠杆菌(革兰氏阴性细菌),金黄色葡萄球菌(革兰氏阳性细菌)和白色念珠菌(真菌)作为实例进行实验。并且在上述优化实验获得的最佳条件下,对新鲜细菌悬浮液(c=108cfu/ml)进行测定。(a)和(b)分别为lps水溶液样品和lps血清样品对该方法的选择性。红色柱表示样品中同时存在竞争分子与lps,黑色柱表示样品中只存在竞争分子。(c)30v电压下三种菌液(108cfu/ml)在的富集荧光强度。
本发明能够检测注射液及脓毒症模型小鼠血清中的lps含量,能快速区分革兰氏阳性菌、革兰氏阴性菌、真菌。
实施例8标准曲线的制备
方法:通过该方法分别测定水溶液和血清中八种不同浓度(50,100,500,103,104,5×104,105,106fm和50,100,500,103,5×103,104,2×104,5×104fm)的lps标准溶液获得线性图。通过测定lps在8种不同浓度的标准溶液获得。每个点对应于三次独立实验的平均值。三次重复实验的rsd<4.0%。
结果:
标准曲线的制备结果如图7所示,从中可以看出,本发明方法的最低检测限(lod)为8fm,线性范围为:50fm-1nm(水溶液样品),见图7(a);50fm-50pm(血清样品),见图7(b);最低检测限(lod)为8fm。
上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的限制,其他的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!